第二节 粘附分子的表达的调节
如前所述,细胞粘附分子不仅具有多种生理功能,在一定条件下也与病理过程的发生密切相关。在细胞因子、炎症介质以及其它因素的作用下,细胞表面粘附分子表达的水平和构型可以发生改变,导致细胞粘附能力的变化。体内某些粘附分子的表达是组成性(constitutive)的,即通常状态下细胞表面就有一定水平的表达,如CD11/CD18、ICAM-1、ICAM-2和L-selectin等粘附分子在相应细胞的静止状态下有一定水平的表达,在某些因素的作用下,这些粘附分子的表达也可发生上调或下调(up-regulation ordown-regulation)。另外一些粘附分子的表达可以是非组成性(non-consititutive)的,即通常状态下这些粘附分子在细胞表面表达很少或不表达,但在某些因素的作用下可诱导表达,如E-selectin、VCAM-1在内皮细胞的表达即属此类。对粘附分子表达的调节有构型调节和表达数量调节两种方式,目前关于粘附分子表达调节的资料大多来自于对白细胞与内皮细胞粘附作用的研究。
一、粘附分子构型改变影响细胞的粘附作用
除了通过增加或降低粘附分子表达水平来调节细胞粘附能力外,某些因素还可以通过改变粘附分子的构型影响其与配体结合的亲和力,从而调节细胞的粘附能力,这使得对细胞粘附作用的调节更为精细和复杂。
(一)LFA-1分子构型改变对其粘附作用的影响
淋巴细胞在受到外来抗原,PMA,抗CD2、CD3、CD44、CD43或抗MACⅡ类分子单克隆抗体的刺激作用活化后,可发生相互凝集,这种凝集作用依赖于LFA-1/ICAM-1的相互作用,而这两种粘附分子在活化淋巴细胞的表达水平并没有显着增加。静止淋巴细胞即表达一定水平的LFA-1和ICAM-1,NK细胞和某些CTL细胞系更是表达较高水平的LFA-1/ICAM-1分子,但它们并不发生凝集作用。上述事实提示在淋巴细胞活化后,粘附分子可能通过构型变化的方式,提高LFA-1/ICAM相互作用的亲和力,从而提高活化淋巴细胞的粘附能力。
1.NKI-L16和活化状态的LFA-1分子 NKI-L16是一种抗LFA-1的单克隆抗体,其识别的表位在静止淋巴细胞暴露的水平很低。当NKI-L16McAb与淋巴细胞表面的LFA-1作用后,不仅不阻断LFA-1介导的粘附作用,反而可以诱导静止淋巴细胞的相互粘附而使细胞发生凝集。这种诱导粘附作用的机理部分是由NKI-L16McAb改变了LFA-1分子的构型,诱导了NKI-L16识别的表位在静止淋巴细胞的表达。NKI-L16识别表位的表达是粘附作用发生的重要条件,但并不是唯一的,因为CTL细胞虽表达高水平的NKI-L16表位却并不发生自发凝集。目前研究认为,LFA-1分子至少以三种形式存在:(1)静止淋巴细胞表达的LFA-1分子,暴露很少的NKI-L16表位,与ICAM-1分子结合的亲和力(affinity)低;(2)中间状态的LFA-1分子,暴露出大量的NKI-L16表位,但与ICAM-1结合的亲和力仍较低;(3)活化状态的LFA-1分子,暴露出大量高亲和力的NKI-L16表位。不同状态的LFA-1分子在淋巴细胞表面的分布方式是不同的,静止淋巴细胞的LFA-1分子分布分散,而活化的外周血淋巴细胞、CTL克隆、效应T淋巴细胞以及活化的CTL克隆细胞的LFA-1分子呈集中分布,在局部形成高密度的LFA-1分子区域,这可能与NKI-L16表位的暴露有关(图2-10,表2-5)。LFA-1分子在局部形成高密度状态可以提高其与配体结合时的亲合力(avidity)。
在integrin家族中,这种精细的构型调节作用并不仅限于LFA-1分子,已发现VLA-4分子同样存在着静止、部分活化和活化三种要构型,活化的VLA-4分子可与VCAM-1和纤粘连蛋白相结合,部分活化的VLA-4分子仅结合VCAM-1分子,而静止状态的VLA-4分子则失去结合任何配体的能力。
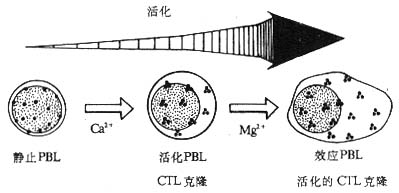
图2-10 淋巴细胞活化后LFA-1分子分布状态的改变
注:静止外周血淋巴细胞(PBL)向活化PBL分化过程中需要Ca2+存在;活化PBL向效应PBL分化以及CTL克隆向活化的CTL克隆分化过程中需要有Cg2+存在。活化的和效应的PBL或CTL表面LFA分子呈集中分布。
表2-5 三种状态LFA-1分子特性的比较
| 静止状态 LFA-1分子 | 中间状态 LFA-1分子 | 活化状态 LFA-1分子 | |
| LFA-1分布方式 | 分散 | 集中 | 集中 |
| 与ICAM-1结合的亲和力 | 低 | 低 | 高 |
| 与ICAM-1结合的亲和力 | 低 | 高 | 高 |
| NKI-L16表位暴露 | 少 | 多 | 多 |
| LFA-1β链(CD18)磷酸化 | 无 | 无 | 有 |
2.Ca2+、Mg2+与LFA-1分子活化状态的关系Ca2+和Mg2+的存在对LFA-1分子与配体的结合是必需的,在粘附试验系统中加入金属离子螯合剂(EDTA或EGTA)去除反应系统中的Ca2+和Mg2+可以完全抑制LFA-1与其配体的结合。采用单克隆抗体对LFA-1分子表位的表达进行检测,发现Ca2+与Mg2+与LFA-1分子某些表位的表达有关,而这些表位的表达是LFA-1分子活化构型的标志。如上述NKI-L16识别表位的表达需要有Ca2+存在;另外一株单克隆抗体24(McAb24)识别的表位在LFA-1、Mac-1和gp150、90均有表达,但依赖Mg2+的存在。PMA或抗细胞表面分子的单克隆抗体作用引起的细胞凝集有一过性持续性两种,一过性的作用在半小时之内消失,而持续性的作用可维持2小时以上。这种现象与离子依赖种类有一定的关系,PMA、NKI-L16、抗CD2和CD44单克隆抗体可以引起持续性的LFA-1分子的活化,它们的作用只依赖Mg2+的存在;而抗CD3、CD43和MHC-Ⅱ类分子的单克隆抗体所引起的凝集是一过性的,它们的作用则依赖Ca2+与Mg2+的同时存在。
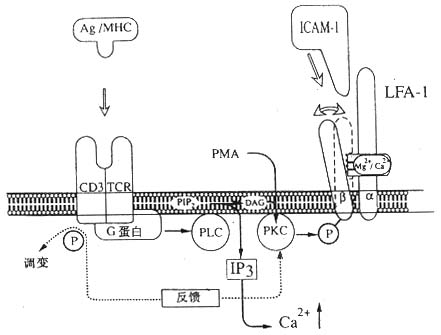
图2-11 LFA-1介导细胞粘附调节的模式图
注:抗原与TCR/CD3复合物结合后激活磷脂酶c,催化PIP2水解为IP3和DAG,引起LFA-1分子β链的磷酸化,使LFA-1分子构型发生变化,提高与配体结合的亲和力。CD3分子的磷酸化引起TCR/CD3复合物的调变,导致PKC水平下降,使LFA-1分子β链去磷酸化转变为非活化状态而产生去粘附作用。
3.LFA-1分子构型改变的机理 目前对于淋巴细胞活化后导致LFA-1分子构型改变的机制还不十分明了。实验表明,PMA作用于淋巴细胞后,通过激活蛋白激酶C(PKC)使LFA-1分子β链发生磷酸化,很可能与LFA-1分子构型的改变有关。抗CD2或CD3单克隆抗体可以通过影响磷酸肌磷酸肌醇代谢途径导致PKC的激活,但两种McAb影响淋巴细胞粘附分子活化的过程是不同的,抗CD2单克隆抗体诱导持久的LFA-1分子活化,而抗CD3单克隆抗体只能诱导短暂的、一过性的LFA-1分子的活化(图2-11)。这种对粘附分子表达的负反馈调节机制,对于体细胞粘附作用的调节过程可能有重要的意义。体内对粘附作用的负调节意味着细胞可以与相互作用的靶细胞脱离,再作用于其它靶细胞,从而最大限度地发挥作用。前面曾提到McAb24识别的表位表达在活化状态的LFA-1分子,McAb24并不阻断LFA-1分子和Mac-1分子与配体的结合,但却可以明显抑制单核细胞向T细胞的抗原提呈作用、LAK细胞对靶细胞的杀伤作用以及中性粒细胞的趋化移动,这些过程均依赖LFA-1和Mac-1分子与其配体的相互作用。单独CD3单克隆抗体只引起一过性的LFA-1分子的活化,而同时加入McAb24则造成持续性LFA-1分子的活化,提示McAb24可能阻止LFA-1分子由活化状态转变为非活化状态。
(二)其它粘附分子构型的改变对粘附作用的影响
除LFA-1分子外,在integrin家族中其它一些粘附分子构型的改变也可以影响细胞的粘附能力。PMA、抗CD2或CD3单抗可以诱导或增强淋巴细胞的VLA-4(CD49d/CD29)、VLA-5(CD49e/CD29)和VLA-6(CD49f/CD29)与其配体(层粘连蛋白或纤粘连蛋白)的粘附作用,提示上述粘附分子可能通过与LFA-1相类似的机制发生构型变化,导致与配体结合的亲和力升高。Mac-1分子(CD11b/CD18)及血小板糖蛋白GPⅡbⅢa(CD41/CD61)分子在细胞活化后可以暴露新的表位,是其分子构型发生改变的直接证据,但其发生机制目前还不清楚。
尽管目前尚未获得selectin家族粘附分子构型变化影响粘附能力的直接证据,但某些抗L-seletin或抗E-selectin分子EGF结构域的单抗非但不阻断L-selectin分子或E-selecti分子与相应配体的结合,反而具有促进作用,提示selectin家族粘附分子中同样存在着分子构型变化对粘附能力调节的可能性。
二、细胞粘附分子表达数量改变对粘附作用的调节
粘附分子表达数量的改变是粘附作用调节的另一个重要方面。粘附分子构型改变与表达数量的增减并不是截然分开的两个过程,两者可能同时存在,共同完成对粘附作用的调节。如淋巴细胞活化后不仅粘附分子构型改变导致亲和力增加,同时也伴有粘附分子数量的增加。
1.调节细胞表面粘附分子表达数量的方式 细胞表面粘附分子表达数量的调节方式主要有诱导贮存在细胞内的粘附分子转移到细胞表面和诱导粘附分子的重新合成两种方式。转移形式的过程发生迅速,只需数秒钟,但维持时间短暂。如凝血酶和组胺作用于内皮细胞可以诱导内皮细胞内贮存在CD62分子迅速转移到细胞表面,然后又很快被内吞而消失;又如CD11b/CD18、CD11c/CD18贮存在中性粒细胞的胞浆颗粒内,在PMA、TNF、IL-1刺激后迅速转移到细胞表面。重新合成过程发生较为迟缓,一般需数小时,但维持时间较长。IL-1、TNF-α作用于血管内皮细胞则可以诱导E-selectin、VCAM-1分子的重新合成与表达,诱导后4小时达到高峰,并可维持24小时以上。
2.细胞因子、炎症介质对粘附分子表达的调节 细胞因子IL-1、IL-3、IL-4、IL-8、PAF、GM-CSF、TNF-α、TNF-β和IFN-γ以及炎症介质白三烯、组胺和凝血酶等可作用于白细胞或/和血管内皮细胞,调节白细胞与血管内皮细胞的粘附作用(表2-6)。在体内可能有多种调节因素同时存在,相互影响,并可能有更多的目前未知的因素参与细胞间粘附的调节过程。
3.细胞的生长、发育状态对粘附分子表达的影响 除了上述细胞因子、炎症介质可以调节细胞粘附分子的表达外,细胞本身的生长、发育、分化及代谢状态也可以影响粘附分子的表达。在胚胎发育过程中,组织细胞粘附分子的表达接一定的规律发生改变,使得不同细胞得以按一定的规律组合在一起,形成不同的组织或器官。肿瘤细胞与其起源的正常组织细胞相比其表达的粘附分子可有很大差异,这可能是某些肿瘤细胞易发生浸润、转移等现象的分子基础。此外,处于不同分化和发育状态的淋巴细胞表达粘附分子也有明显改变,如与未经抗原刺激的T细胞(naive T cell)相比,记忆性T细胞(memory T cell)表达更多的CD2、LFA-1、CD44、VLA-4等粘附分子,而L-selectin在naive T细胞表达水平要明显高于记忆T细胞。
表2-6 细胞因子、炎症介质对细胞粘附分子表达的调节作用
| 炎症介质或细胞因子 | 靶细胞 | 粘附分子表达水平的变化 |
| IL-1 | 血管内皮细胞 | E-selectin↑、VCAM-1↑、ICAM-1↑ |
| 某些肿瘤细胞 | ICAM-1↑ | |
| 中性粒细胞 | CD11b/CD18↑、CD11c、CD18↑ | |
| TNF-α、TMF-β | 血管内皮细胞 | E-selectin↑、VCAM-1↑、ICAM-1↑ |
| 中性粒细胞 | CD11b/CD18↑、CD11c/CD18↑ | |
| IL-3 | 嗜碱性粒细胞 | CD11b/CD18↑ |
| IL-4 | 血管内皮细胞 | VCAM-1↑ |
| IFN-γ | 血管内皮细胞 | ICAM-1↑、VCAM-1↑MHC-Ⅱ类分子↑ |
| PAF、IL-8、 GM-CSF | 中性粒细胞 | L-selectin↓、CD11b/CD18↑ |
| 组胺、凝血酶 | 血管内皮细胞 | CD62↑ |
| 白三烯 | 中性粒细胞 | 粘附作用↑ |
注:↑表示上调(up-regulation)
↓表示下调(down-regulation)