��22�¡���Ѫ�ܲ�����ҩ���ٴ�ҩ��ѧ
��һ�ڡ�Ѫѭ�����ܲ�ȫ��ҩ������ѧ��Ӱ��
һ�����༱֢ʱѪѭ�����ܲ�ȫ�IJ��������ص�
��һ��Ѫѭ�����ܲ�ȫ��ָѪѭ������ҪΪ���ࣩ�����ṩ�㹻����Ѫ�������������֯��л��Ҫ���������༱֢ʱ�ı�ҩ������ѧ����Ҫ�������ԭ��
�������ķθ��ա��ķθ���ʱ����Ѫ�����Լ��٣�ƽ������ѹ���½����ڲ��˴���50%����ʵ�鹷������30%����������ʵ�飬�Ҳ���ɵ�������ͣʱ����Ѫ�������ٲ����ԣ��ĺ����Լ��������ٵ�Ѫ���������Լ��١�
������Ӱ���ҩ������ѧ����
�������������ı������漰������Ҫ������ͼ22-1����
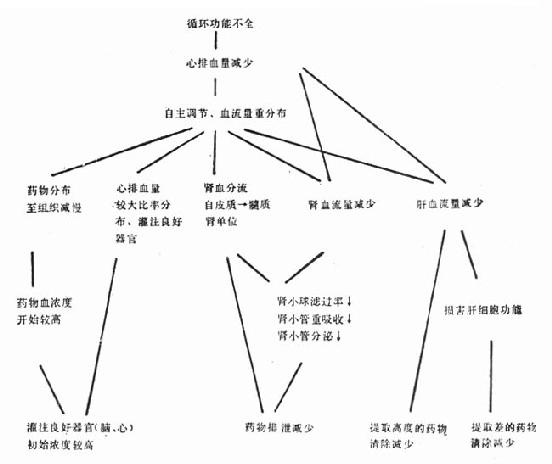
ͼ22-1 ѭ�����ܲ�ȫ��ҩ������ѧ��Ӱ��
��һ���ֲ����� ����Ѫ�����𣬻���Ѫ�����طֲ����ϴֵ�����Ѫ�������ԡ��ĵ����ٵĹ�ע���Ĺ��ܲ�ȫ���ߵ������ҷֲ�������������С����˰�ͨ����������ҩ�ﳣ����������Ĺ���ѪҩŨ�ȣ����ֲ�����Ӧ��
����������� ҩ����Ѫ����������ɾ��ζ�����ת��Ϊ������ʽ������Ի���ԣ���������й���μ������߶������������������������������أ�ͨ�����ٵ�Ѫ���ʺ�������ƵĻ��ԡ���Щ�������Ĺ��ܲ�ȫʱ�����͡�
������������Һ����������ˮ����ҩ��ķֲ��������͵���Ѫ֢�����ӵ�����ʸߵ�ҩ��ֲ�����������ٸ���ȡ��ҩ��ĸ�����ʡ����õ�ҩ��ɲ���Ѫ������ѧЧӦ���Ӷ��ı��ҩ�ﴦ�á�
�������������ڣ�T1/2�������ҩ����һ������ѧ�������������Ѫ���Ƴ���ҩ���뵱ʱ��Ѫ��ҩ��Ũ�ȳɱ������ֲ�������vd���������(cl)�����������ڣ�T1/2�������ϵΪ��T1/2=vd��0.693/cl,��vd��С��cl�ɱ���Ҳ���٣��ɲ��ı�T1/2��������ȸ��踺�������ڼ��ٴﵽ����ˮƽ������˥ʱvd��С������ʹѪŨ�Ƚϸߣ�cl�ӻ���T1/2Ҳ�ӳ��������˥���������ﵽ��̬��ʱ��Ҳ�����ϳ�������ʱ��������С�
�ڶ��ڡ�����ҩ���ٴ�ҩ��ѧ
��Ѫ��ҩ��������Ƽ��ڶ࣬���µ����۰�����ҩ�����á�ҩ������ѧ�������ٴ�Ӧ�á���Ӧ֤������֤���÷������������ͺ�ע�����������Ӧ��ҩ�サ�����á�
��22-1����Ѫ�ܲ���֢����ҩ��
| һ�����Լ���ҩ ǿ��߰������������ë������߰K���ظ��� ��ǿ��߰�ࡡ��Ͱ�����ͷӶ���������ͪ������ͪ ��������ҩ ૱����ᡢ�����ᡢ�������� ����������ʧ��ҩ ������ᶡ����³����˫�����������������������������ͪ�������ɡ��������������ͪ�������а���ά�����ס�����Ʒ�����������ա�����Ӣ�ơ�����������ء����Ρ�þ�� �ġ���Ѫ��ҩ��������ѹҩ�����Ľ�ʹҩ �����ơ�����������������͡��ϼױ����ᡢ��ѹມ���Ѫƽ �塢����ҩ����˨ҩ ���ء�����ø����ø����֯��Ѫ����ԭ����� |
{4}һ��ǿ��߰�����ػ���Digitalis��
���Լ���ҩ��ǿ��ҩ�������ǿ��߰��Ŀǰ���ڼ��ﳣ�õ�ǿ��߰��ë��߰������ë������߰K���ظ�����
��һ��ҩ�����á�ǿ��߰����������ͬ��
1�����Լ������á����ػ����Ƽ�ĤNa+-K+-ATPø������Na+������K+������ϸ����Na+�ѻ�����ʹNa+��Ca2+������Ca2+�������ӣ��ı�ϸ����[Ca2+]�������ļ���������ǿ��߰ʹ˥�����������Ѫ���������������ӣ�����������ĩѹ������ĩ�������ӣ������Ĺ��ܽ����ߵ����ý���������������
���ػ�������Ѫ�����á�������������ǿ�ļ���������֮ǰ�����˹�״����Ҳ���������ã����������ڿ��ٸ�ҩʱ������ҩ������ɱ��⡣
2�����������á����ػ�����������ԣ������ᣬ�ɼ������ʣ�ͨ���������ҽᴫ���ٶȺ��ӳ�����Ч��Ӧ�ڣ��ɼ������ҷ�Ӧ������ʱ�����ж���ǿ��߰�����������ԣ����ƴ����Զ�������������ʧ����
������ҩ��ѧ����ͬ�Ƽ������õ�ǿ���������������в��
��������ë��߰����Cedilanid,Lanatoside-C���ڷ����ղ��10%��40%����Ҫ������ҩ���ظ�����С�����뵰��ϣ���Ҫ������й��80%Ϊԭ�ͣ�17%Ϊ��Ҫ��л����ظ�����T1/2Լ36h��
�ظ�����Digoxin���ڷ�����60%��85%��25%�뵰��ϣ�60%��90%������й��T1/2Ϊ33��36h�������ܲ�ȫʱ�ӳ�����Ҫ�ڷ���Ҳ�ɾ�����ҩ���ڼ��
��ë������߰K��StrophanthineK���ڷ����������գ�2%��5%����90%��100%������й��T1/2Ϊ12��19h��
�������ٴ�Ӧ�á�����ʱ��ѡ�þ�����ҩ��
1����Ҫ��Ӧ֤���ٷ������ˣ������������ߣ��ڼ�������˥��ԭ������˥���أ����۷����������Ķ����٣���ѡ��ά�����ף�W-P-W�ۺ���ʱ���ã���
�÷������������ͼ���22-2��
��22-2��������Чǿ��߰�����˼������÷�
| �Ƽ� | ���ͣ���곣� | ��֢�������ҩ���� | ��Ч�ٶ� | �������ʱ�� | Ч��ά��ʱ�� |
| ë��߰�� | 0.4mg(2ml) | ��ʼ0.4mgС���룬��20ml������ˮϡ�ͺ�����ע����Ҫʱ2��4h���ٸ�0.2��0.4mg | 10min | 1/2��2h | 1��2d |
| ��ë����߰ K | 0.25mg(1ml) | ��ʼ0.25mg����20ml������ˮϡ�ͣ�������ע����Ҫʱ2��4h���ٸ�0.125mg | 3��10min | 1h | 1��2d |
| �ظ��� | 0.25mg(1ml) | ��ʼ0.25��0.5mgϡ�ͺ�ע��4��6h���ٸ�0.25mg | 10min | 14h | 1��2d |
2.����֤���٢�Ȼ��ȷ��Ҵ������ͻ�����Ķ��������ڷʺ��Թ������ļ��������ػ����������ԣ��ɼ��ع��裻��Ԥ���ۺ�������ֱ�ӵ�ת���з�չ������������ʧ��Σ�գ���������������Ӱ��������������ػ����ԡ�ת��ǰӦ��ͣ�ظ���24h�����ػ���߰72h��
���ģ�������Ӧ ���ػ�����ָ���ͣ�������ж�����60%�Բ�������ЧӦ�����¶������ؿ�Ӱ���������ػ������ԣ����������Ƽ���ʱҲ���ܷ������Է�Ӧ����22-3����
��22-3���ػ��ж��״�����
| 1����������� | 3������״̬ |
| ��Ѫ�� | ��״�ٹ��ܼ��� |
| ��Ѫ�� | ��Ѫ��֢ |
| ��Ѫþ | ��˥�ߣ��ظ����� |
| ��Ѫ�� | �ļ��� |
| ���ж� | �½���������� |
| �� | �������༲�� |
| 2��ҩ�� | 4������ |
| ʧ������� | �����ټ����ʣ�����ATPø��ϲ�λ�����ٷֲ��ݻ�������������ʹDigoxin��й���٣� |
| ��θͪ��Carbenoxolone�� | |
| Ƥ�ʼ��� | |
| ��Ѫƽ | |
| ���ᶡ | |
| �����Ӱ� | |
| ����ͪ | |
| ά�����ס�������� |
���壩ѪŨ�ȼ����ٴ����塡�ⶨ��̬���ػ�߰ѪŨ�ȣ��ظ�������6h���´θ�ҩǰȡѪ�걾���������ػ��ж����ж���һ���ο���ֵ�����Ʋ�����ͨ���ظ���ѪŨ��Ϊ0.5��2ng/ml���ڴ˷�Χ��һ��˵ѪŨ�Ȳⶨ��ָ�����������壻��Ѫ���еĺ�����һ���ɿ��ط�ӳ�ļ��ڵĺ�����
��������ҩע������ٸ�ҩǰӦ��ϸ�˽��������ʹ�����ػ��������1�������ù����ػ������ߣ���Ӧ��ͨ����ҩ������Ӧ���������ٺͷִθ��裬�Է��������ڼ����ļ��������1��2�죬���������ػ������ʱ���ļ��ĵ粻�ȣ��׳�������ʧ�����ۼ��ᄇ����ҩ���軻�ÿڷ��Ƽ������õظ�������ĩ����������6h��ʼ��0.125mg q6h�����Σ�������0.25mg��ÿ��1�Ρ�
���ߣ�ҩ�サ�����á�����ҩ����ż�������ҩ�������ĵͼؿ���ǿ���ػ�߰������Ӧ��
���ᶡͬ��ʱ��������ٵظ���������ʺı���֯�ֲ��ݻ�������DigoxinѪŨ�ȣ����Ӳ�����Ӧ������������DigoxinѪŨ�ȣ��Ӷ������ж�Σ���Ե�ҩ�������Ұ���ͪ��ά�����ס�������ड�
������ǿ��߰�����Լ���ҩ
70���������һϵ�з�ǿ��߰�������ļ���������ҩ��½�������ٴ�Ӧ�ã���22-4�����ǽ�������˥��������һ��������½�չ�����������Ӱ���Ķ�Ͱ�����ͷӶ������������Լ�˫�������İ���ͪ��Amrinon��������ͪ��Milrinone���ɾ�����ҩ����Ϊ����ǿ�����ơ�
��22-4ǿ��ҩ�����û���
| ҩ���� | ����Ҫ �����á������� |
| 1�����ػ�ǿ��߰ | Na-K-ATPø���� |
| 2�������Ӱ� | �� |
| ������ȥ���������� | ��1�̼�����cAMP |
| �������� | �� |
| ��Ͱ� | �� |
| ��ͷӶ��� | �� |
| ���DZ��İ���premalterol�� | ��1�˷� |
| Butopamine | �� |
| �ڷ��������� | ��1����2�˷� |
| �洭�� | �� |
| H80/63 | �� |
| �첨������Ibopamine�� | ��1�˷ܼ����֦�1���� |
| 3.˫����� | �� |
| ����ͪ | ��Ca2+�˶� |
| ����ͪ��Milrinone�� | �������ø���� |
| Enoximone | �� |
| 4���������� | �� |
| ����� | �������ø���ơ�cAMP�� |
| 5���� | ��Ca2+�˶� |
| 6���ȸ�Ѫ���� | �̼�cAMP |
�����Ӱ������Լ���������ͨ���˷��ļ���1�������������塣�������Ħ��������˷ܣ�������߰����ø�����ߴ�APTΪ3�䡢5��-cAMP��Ȼ��ø��cAMP���ʹ��Ĥϵͳ���ữ�������ӣ�������Ӹ�������ת������ĸ��������������ö��������Լ���ЧӦ����1�����˷�ͬʱҲ�����ͷźͷ��Ҵ��������Ķ����ټ���������ʧ�����ܳ��֣�����������������Ƽ�������Լ������õķ��ӡ������⽻�а��Ƽ��Ը��ֽ��а���������ò�ͬ����22-5����
��һ����Ͱ���Dopamine��
1��ҩ�����á���Ͱ�Ϊȥ���������ص�ֱ��ǰ�塣��Ͱ��˷����ֲ�ͬ�����壺���˷ܶ�Ͱ����壬С������1��5��g/kg��min������ͻ��ЧӦ��ʹ��Ѫ�ܡ���ϵĤ������Ѫ�����ţ�Ѫ�����ӣ��������ӡ���ֱ���˷��ļ������壨5��10��g/kg��min����Ҳʹȥ�����������ͷţ��������Լ������ã�ʹ����Ѫ���������ӣ����ʽ������ӡ��������ڦ�1���壬����Ѫ��������Ѫѹ���ߣ�С����ʱ��ЧӦ���ԣ�������С��10��g/(kg��min)ʱ������Ѫѹ�������ԣ��ɲ����Ķ����١�����ʧ��������������30��g/(kg��min)������ΧѪ��������ȥ���������أ�����Ӱ����Ѫ������
��22-5���ࡢѪ�ܵĽ��а�������⽻�а��Ƽ�������
| �ܡ� �� | �� �� �� �� �� �� �� | �� �� �� �� �� | |||||
| ��1 | ��2 | ��1 | ��2 | 1 | 2 | ||
| ��λ | ��ͻ�� | ��ͻ�� | ��ͻ�� | ��ͻǰ | ��ͻ�� | ��ͻǰ | ��ͻǰ |
| ЧӦ | �ļ��˷� | Ѫ������ | Ѫ������ | ����ȥ�����������ͷ� | Ѫ������ | Ѫ������ | ����ȥ����������ϡ�� |
| ��Ͱ� | ++ | + | ++ | �� | �� | +++ | �� |
| ��ͷӶ��� | ++ | + | + | �� | �� | + | �� |
| ���DZ��� | ++ | �� | �� | �� | �� | �� | �� |
| ȥ���������� | ++ | + | ++++ | - | ++ | �� | �� |
| �������� | +++ | +++ | +++ | + | ++ | �� | �� |
| ����������� | ++++ | ++++ | �� | �� | �� | �� | �� |
| ���ǰ� | �� | �� | ++++ | �� | �� | �� | �� |
| ɳ������ | + | ++++ | �� | �� | �� | �� | �� |
2��ҩ��ѧ��T1/2Ϊ1��2min���ʴ��������ע���ܵ�������ø���ã�75%��л�������ԣ�25%��л�γ�ȥ���������ء�
3����Ӧ֤����Ͱ������ڼ����ļ���������Դ���ݿˣ�����ʽ������������������ˣ��ڶ�����Ѫ֢��������˥�ߣ���������ΧѪ���������ͻ������IJ��˸����档����Դ���ݿˣ�Ӧ�ڵ�Ѫ����������ʹ�ö�Ͱ���������ʾ�����ڷ�չ����Ѫ�������IJ���������������������ƺ��ã�żҲ��������������غ��á�
4������֤���ȸ�ϸ��������״�ٹ��ܿ������������������洦���ڶ�̥����Σ����ʱ����Ͱ�Ҳ�����������ڡ�
����ΧѪ�ܲ�ʷ���綯��Ӳ��������˨������������ŵ����B��erger���ȣ���Ӧ�ö�Ͱ�ʱӦ���м��֫��Ƥ����ɫ���¶ȸı䣬����֫��ȱѪ��������ʱ����ټ�����
5���������÷�����Ͱ�20mg����5%������Һ��������Һ��100��200ml�����Σ���С������ʼ���������ؼ����Ʒ�Ӧ����Ũ�Ⱥ����ʡ�С������2��5��g/kg��min��ʹ�������ӣ�����Ѫ�������������ӣ��е�����6��10��g/kg��min������������Ѫ��������ά�֣���ʼʹ���ʡ�Ѫѹ���ӣ��ϴ������11��20��g/kg��min��ʹ����Ѫ���������ԣ����ʺ�Ѫѹ���ӣ���ëϸѪ��ѹ���ӣ���������ʧ����Ӧ�ö�Ͱ��������ʵ�����Ѫ�������������ϴ�ͣҩʱӦ���ټ�����ͣ�ã���ֹ��Ѫѹ������
6��������Ӧ ������ļ£�������ǰ�������Ķ����٣������ġ�Ż�¡��Ľ�ʹ��ͷʹ����Ѫѹ��Ѫ���������֣�֫�䡢��ѹ��С�����������Ķ�������QRS������Ѫѹ���ߡ�����Ѫ֢�ȡ�
7��ҩ������á��뵥������ø���Ƽ�����ʱ����Ͱ�����Ӧ���٣�ǰҩ�ɼ�ǿ��Ͱ����ã�����ʼ������Ϊͨ��������1/10��
��Ͱ���ǿ����ҩ���ã�����������Ѫ�ܣ�������Ѫ��������С���˹�Һ�Լ�����й��
��Ͱ�����ȵ���ʹ���á�
��Ͱ���������������ء����������������ƺ��á�������ҩ���Ͱ���Ӧ��IJ��ˣ���Ͱ�������ҩ��������ã��ɲ����Ϻõ�����Ч����
���Ͱ�����ά��Ѫѹ��������ǰ����á�
���ܶ�Ͱ��IJ��˾������豽��Ӣ�ƣ��ɷ�����Ѫѹ���Ķ��������˱�����á�
��������ͷӶ�����Dobutamine,Dobutrex®��
1��ҩ�����á�Ϊһ�ֺϳɵ������Ӱ�����ֱ����ǿ�ļ������ԣ���1�����˷ܣ�������ǿ�����������������أ�����ʱ��ЧӦ����ǿ��Χ����ЧӦ���Խ�С��Ϊ���ŵ㡣����С��7.5��g/��kg��min)������Ѫ�����ӣ����������ʺ�Ѫѹ���ڿɽ��ͷ�ëϸѪ��ѹ�������Ͱ����������߷�ëϸѪ��Шѹ���ۼ�������7.5��g/��min��kg)�����˷ܦ����壬ż�����Ѫ��������ͨ����ΧѪ���������͡�������������Ѫѹ���ˣ��̷�������Ѫ�����͵��жȵ�Ѫѹ���������������Ѫ����������Ѫѹ���������ѹҩ���ã����˵��á���ͷӶ������Ͱ�������˥����Ѫ������ѧЧӦ����ͬ����22-6��
��22-6����ͷӶ������Ͱ�������˥���ߵ�ѪҺ����ѧЧӦ�ıȽ�
| ����Ѫ�� | ����ѹ | ��Χѭ������ | ���ҳ�ӯѹ | ���� | ÿ����Ѫ�� | �IJ�����ָ�� |
| ��ͷӶ������� | �� | ���� | �� | ���� | ���� | ���� |
| ��Ͱ����� | �� | ���� | ���� | �� | ���� | ���� |
����ʾ���䣬�� �����������ʾ���Լ��ٻ����ӡ�
2��ҩ��ѧ��1��2min����Ч����߷�����һ����10min��Ѫ��T1/2Ϊ2min����Ҫ��л;��Ϊ�����Ӽ����ͽ�ϡ�������Ҫ��й����Ϊ��ͷӶ�����3��0������Ͱ���������ȩ���Σ��������ԡ�
3����Ӧ֤���ļ��������������³�������˥�ߣ���Ϊ���Լ���֧�����ƣ��ɾ�����ʱ���裨24��48��72h���������ļ���������ӦС��Ӧ�á�
4������֤���ʺ����ļ������ķ�����Ӧ�������ػ����������ʣ�ԭ�и�Ѫѹ������Ѫѹ�������ߣ�������̥��������δ��ȷ��
5���÷����������Դ�������ˣ���ͷӶ���2.5��10��g/��kg��min)���ɻ����ⷴӦ����һЩ����0.5��g/��kg��min)��Ч����һЩ��40��g/��kg��min)���������Һ�ÿ��Ƹ�ҩ��
ÿСƿ����ͷӶ���250mg��100mg���ɼ���5%������Һ��������ˮ��N/6��������Һ�У���Һ�ʷۺ�ɫ����ʱ������˵��ҩЧ������ʧ���ɰ���22-7ϡ�͡�
��22-7��ͷӶ������Ʋο���
| ���Ũ�� | 100mg | 250mg |
| ������Һ�� | ������Һ�� | |
| 1000��g/ml | 100ml | 250ml |
| 500��g/ml | 200ml | 500ml |
| 250��g/ml | 400ml | 1000ml |
| 200��g/ml | 500ml | �� |
��ҩ�ڼ�Ӧ��������ĵ�ͼ��Ѫѹ������ܼ��ζ���Шѹ������Ѫ������ҩǰӦ���ʵ�����Ѫ�����Խ�����Ѫ������
6��������Ӧ���������ġ�ͷʹ���Ľ�ʹ������������ʹ���������������죬Ѫѹ����������ѹ�����ߡ���������λ���ɣ��緿�硢���磬ż�������Ķ����١�������Ӧ��Ƥ����ߡ����ȡ�������ϸ�����࣬ż��֧���ܾ��Ρ�
7��ҩ�サ�����á���Ӧ����������ͼ����ĵð����ã�����߿ɸ��������á���ͷӶ����������ػ����������ࡢ����ҩ�������Ⱥ��á�
��������ҩ
����ѡ�õ�����ҩ��Ҫ������Ѹ�١�ǿЧ���ڸ�������ҩ������������ҩ�������ᡢ૱����ᡢ��������ȷ�������Ҫ��
��һ��૱����ᣨ����Furosemide,Lasix��
1��ҩ�����á�������Ѹ�١�Чǿ������Զ��ݡ���Ҫ��������С��������֧���ʲ�������Cl-���������գ�Na+������Ҳ��֮���٣���Na+����Cl-��Ҳ��K+�����µ�Ѫ�ء����������ã�����ɴ̼����ط��ڣ�������Ƥ��Ѫ�ܣ�������Ѫ�����������ܲ�ȫҲ���ã���������������������Ѫ�ܴ���������ҩ�ɸ��Ʒγ�Ѫ�����������ҳ�ӯѹ������Ѫ������ѧЧӦ����������֮ǰ��
2��ҩ��ѧ����ע����30min��߷壬�ڷ���Ϊ1h�����ó���4��6h���ڷ������յ�����ȫ���������95%��99%�����չ�̥�̺�����֭���ڡ�������й����Ϊԭ�ͣ�10%��л����������ȩ���ϣ�Ҳ����֭��й��T1/2Ϊ30��70min����˥����˥ʱ�ӳ���
3������Ӧ�� �����ڼ��Է�ˮ���Ѫ������˥�ߡ���Ѫѹ��֢�������ۺ�������Ӳ����ˮ�ס�����������ҩ��Ч����������Ч��
���ﳣ��ע������ע��1��2min��������20��40mg�����ٵ�1��1.5h��������ٸ���2������������ڼ��Ի�����������˥�ߣ���ʱ������1000mg/d��10�����ϵij�����ҩ������ЧӦ�������˼�Ъ�Ʒ�����ҩ1��3�죬ͣ��2��4�졣ÿ֧20mg(2ml)��Ƭ��20mg��
4��������Ӧ����Һ��͵����ʧ�⣺���������Ѫ֢���ͼ�Ѫ֢����þѪ֢����Ѫ�����ɰ��Ѫѹ���ɳ��ֵ�������ҵ�һϵ��֢״����ʳ�������ġ�Ż�¡�ƣ�͡��������ڿʡ�ͷ�Ρ������εȡ���������л�ı䣺������Ѫ֢����Ѫ��֢���������Եȡ��۲������IJ�����Ӧ���������ס������Ի��ԡ������Ƥ�ס�Ƥ����ߡ��������ơ�����ϸ�����١�ѪС����١���ʱ�Զ������������ˣ�ѪҩŨ�ȴ���50��g/ml����
5��ҩ������á���������������й���ù�ء�ͷ�߾��غ͵ظ���������ǰ���ߺ���ʱ�������������Ͷ����ԣ���������˥��ʱ��������ø���������������ĵ�Ѫ���������ػ��ж��ķ����ʺ�Σ�ա�
����ɸ���ˮ��������й�����ߺ���ʱ����ʱ�ͼ������ɳ���ˮ�����ж���
�����3������ñ���������ǰ���ٷʴ���������Ѳ��������á�
����Ӧ��ǿ������Һ��ϡ�
�����������ᣨEthacrynicacid,Edecrin��
1��ҩ�����á�������Ϊ������ҩ������ǿ��
2��ҩ��ѧ���������ã���ע10min���ҳ��֣�����2h���ڷ�30min����Ч��2h��߷塣���ó���6��8h��95%Ѫ������ϡ��ֲ��ڸ�Ũ�Ƚϸߣ���Ҫ�ɽ���С�ܷ��ں;���֭��й����������������á�
3������Ӧ�á����Ƹ���ˮ�ס�ÿ֧����������25mg�����Է�ˮ��ʱ����25��50mg������������20��40ml5%������Һ��������ˮ��������ע���㡣
����ע�ⲹ�ء�
4��������Ӧ�����������������ң�θ����֢״��������ʳ�����ġ�Ż�¡���к������θ������Ѫ����ѣ����ʱ�Ի������Զ����������ɳ����ھ�ע100mg��1h�ڣ�Ӧ�����백����߰����غ��á��������˿��¸�ϸ������ϸ�����١�Ƥ��ȡ�
�������������ᣨ����Bumetanide,Bumex��
1��ҩ�����á�Ϊ���µ���������������û�������������ЧӦ������ǿ20��40�������������С��ʧ�����ý������ᡣ��������Ѫ�����á�
2��ҩ��ѧ���ɾ���ע�䣬�����Ӻ�ʼ����30��60min��߷壬���ó���2��4h��T1/2Ϊ1��1��1/2��h���ڷ�����Ѹ�٣�30min��Ч��1��4h��߷壬����4��6h��������ʴ���95%���ֲ��ݻ�0.2��0.3L/kg�����ִ�л��������ԭ�������ų���
3������Ӧ�á����ڸ��������ˮ�ͷ�ˮ�ס���������˥�߿ɾ�ע��ע0.5��1mg��ÿ֧2ml��0.5mg������Ҫʱ30min�ٸ�1�Ρ��ڷ�1��10mg/d���ִθ���ÿƬ1mg����
4��������Ӧ��ͬ�����������ʵ͡����˼���������Һ�о��㣬�Ա��ⷢ��������
�ġ�������ʧ��ҩ
Ŀǰ�ٴ�Ӧ�õĿ�����ʧ��ҩ�����Ǹ���ҩ����Ҫ���������ö����ֵģ���22-8����
�ڢ�����ͨ�����ͼ���Ҳ��Ĥ���Ƽ����о������ã����ļ�ϸ��Ĥ��λ������ֱ���������ã������п����������ã�ͨ��ֲ�����Ӱ�촫��ϵͳ�������������Զ�����λʱ�䣬4�������λ�¶�Ҳ�в�ͬ�̶�Ӱ�죬������Ӱ��̶��ֿɷ�Ϊ��A����B����C�������͡�
��22-8��������ʧ��ҩ���ࡪ���������û���
| ���� | ���� | ���� | ���� | ���� | ���� | ||
| A | B | C | |||||
| ��Ҫ���� | ��ͨ�����ͼ� | �������� | �ӳ����� | ��ͨ������ | �� | ||
| ����O�� | ����O�� | ����O�� | �� | �� | �� | �� | |
| �ж� | �� �� | �� �� | �� | �� | �� | �� | |
| �������� | �������� | �������� | �� | �� | �� | �� | |
| ++ | 0��+ | ++++ | �� | �� | �� | �� | |
| �ӳ����� | ���̳��� | �Գ����������� | �� | �� | �� | �� | |
| ҩ�� | ���ᶡ | ����� | Ӣ���� | �����ͼ� | ����ͪ | ά������ | ���ػ� |
| ��³���� | ����Ӣ�� | ������ | ������� | ���а� | ��
| ���� | |
| ˫������� | �����ױ��� | �ȿ��� | �����İ��� | �� | �� | ����������� | |
| �� | ������ | �������� | �� | �� | �� | ϩ������ | |
| �� | ������� | ��������ͪ | �� | �� | �� | �� | |
| �� | �� | ������������ | �� | �� | �� | �� | |

���ﳣ�õ��������
���۴˴�ȱ��һЩ���ݣݡ�
��Ӱ��С�������Ƽ����������������֢״�Ի��ٴ���Ҫ��Ѫ������ѧ�쳣���ɰ�ȫ���������ػ�������˥���ߡ���������Ӱ��������ϵͳ��С����������������ʹ�����������ã���������������ʼ�����ֹͣ��
2��ҩ��ѧ�������ڷ�������գ�������߶ȵĸ�����ʣ���ֻ�˾�����ע��ҩ�����������3min�ڼ����Ũ�ȣ�����10��20min���ʶ���ģ�ͷֲ������������1��2h�����������ʧ����Ч����ѪŨ��Ϊ1.5��5��g/ml(��6.5��21��mol/L)��Ũ��Ϊ3��5��g/mlʱ�����������¶����ý��棬����6��g/ml(25��26��mol/L)�������������ж�֢״������˥�ߡ���Ըβ�ʱ���������ʽ��ͣ�������ӳ����׳����ж�֢״�������24h�������Ҳ�ӳ����ɴ�4h�����˼���ʹ�á�
3����Ӧ֤���ٸ��������粫���ر���Ƶ����5��/min���ϣ��������ɻ��Դ�ԣ�������T�����壻���ּ���������缱���ļ��������ĵ��ܼ������������ʱ����Ч�ѣ��������Ķ����٣�Ч�ѣ������ػ��ж���縴�ɺ�����Կ���������ʧ�������Ҳ����������ֹͣ��Ч�ѡ�
4������֤���ٶ�������ҩ�����ߣ��ڸ߶������һ����������ͣ�������Ķ����������ԣ����ҽ����ԣ��ݲ�������Ԥ���������������ʹ�������죬���������������ݲ�Ƶ�ʣ�����ָ����ص���������ʧ�����ܷ���������Դ�����QRS���Σ���������������ӷ��Ҵ��������������ʡ�
5�������÷�����̣�ÿ֧0.2g(10ml)��0.4g(20ml)������С���룬���μ���Ϊ��50��100mg��С���루��ϡ�ͣ�����ÿ15min50mg����Ҫʱ�ظ�1��2�Σ�ͬʱ���Σ�����1��3mg/min(��100��300mg��������5%������Һ100ml��)��ÿ����1ml���ú�����Һ�õ��ڡ�
6��������Ӧ��������йأ�ͨ�������ڼ�����200��300mg/h����ʱ���ֲ��ɷ���Ѫ˨�����ס�
��ϵͳ����ͷ�Σ����������죬��˯���������������ˣ�����ģ�����ӣ�������˵�����������ѣ��ȡ������о���Ż�£�����������ֲ���ȫ���鴤�������ʣ���־���壬����������������ֹͣ��
��Ѫ��ϵͳͨ������Ӱ�죬������ʱ�ɲ�����Ѫѹ���ݿˡ��Ķ���������ȫ�Է������͡����ͻ�����ͣ�١�
�緢�����ط�Ӧ��Ӧ����ֹ��ҩ�����д��������ó������õİͱ����Σ�����������0.1��0.2g��10mg��ע��
7��ҩ������á����������³��������������������ᶡ��Ľ������к��������ɷ��������������³����ͬ�ã������������������ԣ��������겻�������ӻ�����֢״��
�ĵð�������������ԡ���������Ҳ������������ԡ�
���������ᶡ��Quinidine��Ϊ�����������塣
1��ҩ�����á�����A�����ʧ��ҩ����Ҳֱ�����ô���ϵͳ������AV���������ӳ�P-Q���ڣ��ӳ����Ҽ�������λʱ�䣬������QRS��QT���ڣ�QRS�϶���ֵ����25%��50%Ϊ����֢״�����п����������ã��ɼӿ������ʣ��ر��ڷ�������ʱ�����Ц������������ã����������ಡ������������ʧ�����Ѫѹ�����ᶡ���ļ��������á��ܦ������������ã�ʹ��ΧѪ�ܶԦ������������˷ܼ�����Ӧ���ʿ��ᶡ����ĵ�Ѫѹ���ء�
2��ҩ��ѧ���ڷ��������á��ڷ����������ʱ��1��3h�����ó���6��8h��T1/2Ϊ6h,�������ӳ���9.7h���ң���80%��Ѫ������ϣ���������;��������10%��20%�Բ�����ʽ�����ų�����Ѫ����˥�������ܲ�ȫʱ�����ᶡ����й������Ѫ��������Ũ�Ƚϸߣ��˼��ټ������������ӻ����ᶡ��й���������������й�����Ʒ�Χ��Ѫ�����ᶡŨ��Ϊ1.5��5��g/ml(ƽ��3.5��g/ml)���˲��ֵ��ֵ��ĩ�μ�����1h��6��8hȡѪ����Ϊ���ᶡ������ȩ���Σ���ֵ�ڿڷ���4��8h���������ܲ�ȫʱ�����ᶡѪŨ�ȿ����ߣ���������ʾ���ᶡ���ԡ�
3����Ӧ֤���ٷ����粫��Ч�ѡ������Է����Ķ����٣�Ч�ѡ��۷���ת����ά����ɣ�Ч�ѡ�ת�����̿ɳ�����ʱ�Է��ˣ�����ҩ�����з��˳�����Ӧͣ���ᶡ����ΪԤ�����������������ö����������ʣ����ȸ���Ч���ػ�����Ԥ���ۺ����ϲ����Է�����Ч�ѡ��ݷ���ת�������ػ�����Ч�ɡ��������粫��Ч�á��߽����������Ķ����٣�Ч�á��������粫�������Ķ����٣�Ч����Ч�ѡ������ػ����������Ķ����٣�Ч�ɡ�
4������֤�����Ҳ������ᶡ�����Ҳ������ȣ����ӵ縴�ɵ��ܣ��ڢ�Ȼ���ȫ�Դ������ͣ��۶Կ��ᶡ�������ҩ�����ز�����Ӧ����ʷ��
5���������÷���������ᶡ��ÿƬ0.2g����Ҫ�ڷ���Ϊ����Ŀ�ġ���һ��0.2g��ÿ2hһ�Σ���5�Ρ�����ЧҲ���Է�Ӧ����2������0.3g��ÿ2hһ�Σ�����5�Σ�ÿ������һ�㲻����2g��ÿ�θ�ҩǰӦ��Ѫѹ���ĵ�ͼ���ָ��������ɺ���ά������һ��Ϊ��Ч����ȥ0.2g��2��3��/d�����ᶡ��Ч�������ж�����֮���������խ�����˾����ÿ��ᶡ��
6��������Ӧ ���������ӦΪ���ġ�Ż�¡���к��ͷ�λ�ͷʹ�������������ϰ������ɷ�Ӧ�����粻����һ��ɲ�ͣҩ���䷢����ѪҩŨ������ء���Ѫ��ϵͳ�����Ķ�����������˥������Ѫѹ���ݿˡ���ȫ�Է��Ҵ������͡�����ͣ����Ťת�������Ķ����١��Ҳ��ȡ�����Ϊ���ز�����Ӧ�����ڼ��Ťת�������Ķ����٣�������������ᶡ���������ɷ���������ѪŨ�ȷ�Χ�ڡ�������Ӧ��Ѫ������ˮ�ס������ʷ�Ӧ��������������ѡ�����ȡ�
7��ҩ������á����ᶡ������С�ܶԵظ�������й��ʹ�ظ�����̬ѪŨ�ȱ��������ߺ���ʱ���ظ�����Ӧ���롣���ͱ��ס�����Ӣ������ƽ��������ᶡ�Ĵ�л���������ʱ���ᶡ���������ᶡ�������ӳ�QT���ڵ�ҩ�����³��������ͪ��˫��������ȣ����á������������ĵ�Ѫ�ؿɼ������Ϳ�����ʧ��ҩ��ЧӦ��Ӧ�ÿ��ᶡʱӦ�Ⱦ�����Ѫ�ء�
��������³������³����������Procainamide,Pronestyl�����PA��
1��ҩ�����á�����A�Ϳ�����ʧ��ҩ���ƿ��ᶡ�������ļ������ԣ��������Ҵ����ٶȣ��ӳ�������λʱ��Ͳ�Ӧ�ڣ��������������������á��ɷ�����Ѫѹ���ɼ����ļ���������������������������ĩѹ��Ҳ��������Ѫ�������߷ζ���ѹ��
2��ҩ��ѧ���ڷ�����ȫ���ա��������ö�Ϊ������75%��95%���ڷ���1h����ע��30min��ѪŨ�ȴ�߷塣�������������Ͽ���ʽ����ģ�͡�ƽ��T1/2��Ϊ5min��T1/2��Ϊ3h(2.2��5.0h),���������ܲ�ȫ�����ӳ���PAԼ50%�ڸδ�л�������������γ���Ҫ��л����μ�����PA��л������Ӧ��СPA��������Ѫ����˥����ҲӦ��Ӧ���ٸ�����ά�ּ���������ѪŨ��Ϊ4��8��g/ml(ռ85%����)����10%��Ч����ѪŨ��Ϊ8��12��g/ml��
3����Ӧ֤���������Է��Ի���������ʧ�����ر����������ʧ������Ч���������粫�������Ķ�����Ч�ѣ��������ػ����������ٲ���AV���͡�����Ч�á�
4������֤����Ȼ���AV���͡���֢��������������Ӧ�ߣ�����³������ܲ������������Ӧ����
5���������÷��������������þ�ע�Ρ���עΪ3��5min�ھ�ע100mg��ÿ��5��10min�ظ�һ�Σ�ֱ����Ч��������������10��15mg/kg��Ҳ�ɾ��Σ�0.5��1g����5%��10%������Һ100��200ml����ʼ10��30min�ٶȿ��ʵ��Ͽ죬��1h�ڵ��꣬��Ч�ߣ�1h����ٸ�1�Ρ�����������2g/d��
6��������Ӧ���ٶ��ġ�Ż�¡���ʳ���ڷ�ʱ��������������е�Ѫѹ�������ݿˣ����侲����ҩʱ���ɷ������ڴ������͡�QT�����ӳ��������Ķ����١����Ҳ�����ͣ������˥�ȡ����������ʲ��˿ɷ���������Ӧ���ɼ�Ƥ����ȡ�����ʹ����ϸ�����١�Ѫ������ˮ�ȡ����Դ���Ϊ����ͣҩ�����ص�Ѫѹ��ʱ������ѹҩ������QRS���������ʼ������ɾ�������̼�����ơ�
7��ҩ������á������ĵð����ã���ҩ�����˼��١��������ػ����á��������ӳ�QT���ڵ�����ҩ������ᶡ�������¡�Ⱥ��á�������ҩ����ѹҩ����ʱ�ɼ��ص�Ѫѹ���á�PA����ǿ�������ͼ����ã�����ʱ�ɲ������غ������ơ�
���ģ�˫���������Disopyramide,Rythmodan,Norpace��
1��ҩ�����á����ƿ��ᶡ�����Т���ֱ������ϸ��Ĥ���ã�����a�ࣩ�����̡�����Vmax������4������¶ȣ��ӳ�������λʱ�䡣���н�ǿ�Ŀ����������á����и��Լ������ã�������������Ѫ����������Ѫ�����Ĵ������ܽ���ʱ���շ�����˥�ߣ��������ھ����ø�����ʱ��
2��ҩ��ѧ�������οڷ�����Ѹ�١���ȫ��90%������Ũ�ȳ�����2��3h��Ѫ���������50%��80%�����۷ֲ��ݻ�0.5L/kg��Լ50%��ԭ�������ų������־�N-���ǻ����ô�л����л�����ͻ���Ŀ����������á�T1/26��10h�������ܲ�ȫʱ�ӳ���ͨ����Ч����ѪŨ��Ϊ3��5��g/ml���䵰�����ΪŨ�������ԡ�����ѪŨ�ȶԲ�����Ӧ�����Թ�ϵ��Ӧ��ʱӦ���������ѪŨ�ȡ�
3����Ӧ֤�����������Ժ�����������ʧ�����������粫�������Ժͷdz����������Ķ�������Ч�����ڿڷ�����Ч��50%���ϡ�����Ȼ���������٣���ע��75%��95%��ֹ��������20%��66%����̼��շ���������Ԥ�����á���������Ч���ϲ
4������֤����Դ���ݿˣ�ԭ�Т�Ȼ��ȷ��Ҵ������ͣ�δ��װ�����ߣ�����֪�Ա�ҩ�����ߣ�����ۺ�����ǰ���ٷʴ��������˥�����á�
5���������÷���50��100mgϡ�ͺ�ע��5��10min�ڻ���ע�꣬��Ҫʱ45��60min����ٸ�1�Σ����Σ�100��200mg��5%������Һ500mlϡ�ͣ��ٶȲ���̫�죬һ��0.5��1mg/kg��h����20��30mg/h)��ÿ֧50mg(5ml)���ڷ�100��150mg��ÿ��3�Ρ�ÿ������Ϊ100mg��150mg�����ͽ��ң�Norpace CR��Ϊ100mg��150mg��ÿ12hһ�Ρ�
6��������Ӧ���ٿڸɡ�����ģ�����������ѳ������������������£�������Ҫ������������棬�ɲ�����������˥����Դ���ݿˣ����Կڷ���Ҳ�ɷ�����ż����������������Ķ����١��Ҳ�������ǰQRS������QT�����ӳ�����ʼ��ҩ��ӦEKG��⣬һ������QRS������Ӧͣҩ����θ�����ɷ������ġ�Ż�¡���к�����иι��������ֻ��㡣����ϵͳ��ͷ�Ρ��������뵡�ȼ��Ծ���֢״�����������Ѫ�ǡ�
7��ҩ������á���������ʧ��ҩ����ʱӦ����������ã�����QRS������Ӧͣҩ������������ͼ��ĵð��Ⱥ��ã��ɼ��ظ��Լ������ã��շ���˥����ظ������ÿɸ��Ʊ�Ʒ���Լ������á��뱽��Ӣ�ƺ��ã��ɼ��ٱ�Ʒ��л��Ӧ���������
���壩�����������������ɶ���Aprindine,Aprinidine��
1��ҩ�����á�����C�����ʧ��ҩ������Ӱ���С�������ķ������Ҵ������ӳ�A-H��A-V���ڼ��䲻Ӧ�ڡ������ֿ�Ұ��ά������λʱ�䣬ʹ��Ч��Ӧ�����̣�����Ϸ�����·�������ã����ӳ���·��Ӧ�ڡ�����Ӱ��ֲ����ϵͳ��������ҩʹ��
���۴˴�ȱ��һЩ���ݣݡ�
50mg��ÿ��3�Σ��Ժ�ÿ��50mg��ÿ��2�Σ���ÿ��25mg��ÿ��2��3��ά�֡�Ҳ�б����״�100mg����Ѹ�����ѪŨ�ȣ��Ժ��ٰ�һ��������á�ÿƬ25mg��50mg������ʱ�״�100��200mg����5%��10%������Һ100��200mlϡ�ͣ�����Ϊ2.5(2��5)mg/min����Ҫʱ30min��6h�ɷֱ��ٸ�100mg��24h����������300mg�����ո�Ϊ�ڷ������ﲡ�������ĵ�ͼ����µ��ٿ�����Ϊ10mg/min�����й۲�QRS��������15%Ӧ�������٣����農��ά�֣�Ҳ�����ռ��������50��100mg/dΪά�ּ�����
6��������Ӧ����Ч����Ũ�ȷ�Χ��խ������������Ӧ����������ϵͳ��Ӧ��ѣ�Ρ��о��쳣�����������ӡ��þ�������ʱ�ɳ���������鴤��ͣҩ3��4��ɻָ�����θ������Ӧ���ۼ��������ע���ٶȹ��������QRS������P-R��������ӳ���ͣҩ15��20min���ָ�������ʱ�ɳ��������Ķ����ٻ��Ҳ�����Ҳ�г�����ϸ������֢�����ڳ�ʼ4��6�ܳ��֣��ͻ���ı�����Ӧ���ڸ���WBC���ι��ܡ�
��������������ͪ������ƽ��Profafenone,Rytmonorn��
1��ҩ�����á�������C�࣬Ϊ��������ҩ������ϸ��O�������ٶȣ�Vmax��������ӳ�������λʱ�����Ч��Ӧ�ڡ�һ�ξ�����ҩ��A-H��H-V���ķ���������Ч��Ӧ�ڡ�A-V����ʱ����ӳ���QRS��Ⱥ��P-R����������������������Χ�ڣ���������������������������������ಡ�йء�QT�����ӳ�20%���ң�˵������������ϵͳ�����������ã�Ҳ�ӳ���·������������������ļ����ã���������ĩ��ѹ�����ٲ���������������������
2��ҩ��ѧ���ڷ����������ã�2��3h��߷�ѪŨ�ȡ��ʶ���ģ�͡������ƽ��3h���������ڴ�л��ǿ��֮�֣�T1/2����10h��Ϊ����л�ͣ���Ϊǿ��л�͡�����ѪҩŨ�ȸ����������Ӽ�����ѪŨ�ȳʷ�����������
3����Ӧ֤���������Ժ���������ʧ�����нϺ���Ч��������80%���ϲ�����ȫ���ƣ������Ķ����ٿ�����90%���ϡ����ַ����������Ķ�����Ч���ȷ����Ķ�����Ϊ�ã����ٿɼ��������ʡ������Է��������ˡ�������ת��������WPW�ۺ����鷿��ʱ�������Լ��������ʣ�����Ԥ���ٷ���
4������֤ ��������˥�ߡ���Դ���ݿˡ�����������ۺ��������Ե����ʧ�������������Էβ����ߣ����Ե�Ѫѹ���á�
5���������÷�����ע����Ҫʱ��ECG��½��У�70mg(ÿ֧70mg/20ml)��3��5min��ע�룬���10��20min���ٸ�һ�Σ������������350mg��Ҳ�ɰ�0.5��1mg/kg��ע�Ŀڷ����ڷ��״���300��600mg���Ժ�600��900mg/d����2��3�η��á�Ƭ��ÿƬΪ50mg��150mg��
6��������Ӧ�����١��ٶ��ġ�Ż�µ����θ������Ӧ������ζ���ϰ�����������ͦ��������ã��˶������½���������Ѫѹ���ͣ�P-R�ӳ��������������ò����ĵð���1/4�����ڷ�ҩ��1��2h��ʧ�����б��������Ĺ���������˥�ߣ����������֧�ͷ��Ҵ������ͣ���������ʧ��������Ƶ����
7.ҩ������� ��ظ���ͬ��ʱ����Ʒ��ʹ�ظ���ѪŨ�����ߡ�
���ߣ������ɣ������ɣ�Mexiletine��
1.ҩ�����á����������IB�Ϳ�����ʧ��ҩ���ɿڷ��������ļ��췴Ӧ��Ĥ��Ӧ�ԣ����̶�����λʱ�䣨APD�����ӳ���Ч��Ӧ�ڣ�ERP����ERP/APD��ֵ����ֲ����ϵͳ���������á�
2.ҩ��ѧ���ڷ��������öȸߣ���90%��2��4h��ѪŨ�ȸ߷壻�ֲ��ݻ�9L/kg����Ѫ����������жȣ�70%��������T1/29��12h����Ч����ѪŨ��0.5��2��g/ml��Լ10%������й��
3.��Ӧ֤ ���ƣ�����Ԥ�����Կ���������ʧ�����缱���ļ�����ʱ��������Ѵ���QT�ӳ��ۺ��������ػ��ж��ȣ����Է��Կ���������ʧ��������Ч��
4.����֤ �ж����ϴ������͡�����ۺ����������Ķ�����������˥��ʱ���á�
5.�������÷�����ע��70��100mg�����ÿ֧100mg/2ml������5%������Һ20ml��������ע��3��5min��������Ч����10��20min�ظ�һ�Σ��Ժ���1.5��2mg/min������3��4h�����ٶ�0.75��1mg/min����ά��24��48h���ڷ���Ƭ��ÿƬ50mg��100mg��һ��ÿ��150mg��ÿ6��8hһ�Σ���Ч�����������ÿ��100mg��ÿ��3�Ρ�
6.������Ӧ ������ϵͳ֢״��ͷ�Ρ���������ӡ����ߵȣ�������֢״�ж��ġ�Ż�£���Ѫ�ܷ�Ӧ�е�Ѫѹ���Ķ����������Ҵ������ͣ����侲����ҩ��
7��ҩ������� ���������Կ�����ʧ��ҩ�ɼ�ǿ��ЧӦ��������������ͼ����ظ�����ȫ���ã�������ֹʹҩ���ӻ������ա�
���ˣ������������ĵð���Propranolol,Inderal��
1��ҩ�����á�Ϊһ����õĦ��������ͼ���������Ц�1�������������壬��ȥ���������ء��������أ�������������أ���Ӧ������������Լ������ã���������ǿ�ȣ������Ա�ʱ��ЧӦ���������ʣ����ĵð����ļ������ഫ����֯�����岿λ�������Ӱ����о���������������Լ������Ա�ʱ����ЧӦ��Ҳ������С���������ն�����Ѫ����������ˮ��������Ѫ����˥֢״����
�ĵð����з������Կ�����ʧ�����ã����������ʧ��ҩ���俹����ʧ��������Ҫͨ�����Ͷ����λ�����������ܼ������ã�����Ӧ���Ժʹ����ԣ���ʹ�������ȴ̼������ڼ����ļ����������ػ��ж�ʱ����������ʧ���Ԧ��������ͼ���Ӧ���ã����������ͼ�Ԥ���ļ�ȱѪ���ɼ��������ԣ������۷����ơ�
�ĵð��ĸ��Լ������ÿ�������������ĩѹ���ߺ�����Ѫ�����ͣ��ɱ����ػ����ֵ��������ĵð����������������ҩ�����Ѫ�����ã����ɽ���Ѫѹ��
2��ҩ��ѧ���ڷ��������ã��Ρ�������л�������������ö�ͨ������40%����Ũ�ȳ����ڿڷ���1��4h���ո�ʱ���ʳ�����ո��죬���ó���5��6h���ĵð�����ѪŨ��30��100��g/ml�����ڷ���1h��1��8hȡѪ���ֵ����ֵ����ѪŨ�ȴ���120��g/ml��������ʧ������δ�ﵽ����Ӧ�ټӼ�����Ӧ��ҩ�������壬�ĵð�85%��95%��Ѫ������ϡ�
������ҩ�����ЧӦ������10min�ڣ�����Լ1h?
���۴˴�ȱ��һЩ���ݣݡ�
mg��ÿ��3�Σ��ȸ�ϸ������ǰ3���ҩ��ÿ��10��20mg��ÿ��3�Σ����Ľ�ʹ��ÿ��10mg��ÿ��3��4�Σ�������80mg/d�����
6��������Ӧ����Ҫ�Т��Ķ�������������˥�ߣ��շ�����أ�����֧�����������շ�����أ����ܷ�����˼˯��ͷʹ��ʧ�ߡ����ġ����͡����صȣ�����ΧѪ������������������ŵ��������Ӧ���ĵð�������ͻȻͣҩ���ɳ����Ľ�ʹ���أ������ļ���������������ͣ�á�
7��ҩ������á��ĵð��Ȧ����ͼ���������������ʧ��ҩ���ã��������ػ��Ƽ����ã������ڼ�ǿ���Ʒ��������˵������ʺͼ��٦����ͼ��ĸ��Լ������ã�����ᶡ����³������������ʱ���С�������Է���ǿ�ļ��������á�ԭ����Ѫƽһ��ҩ�������ĵð����ܲ������Ƚ������ͣ������ĵð����ܼ�ǿ����������ҩ��������ҩ��ֹʹҩ���ͱ����ࡢ������ҩ���Ҵ��ȵ����á��������������ĵð��δ�л���������ĵð�ѪŨ�ȣ�ά�����ȸ�����������ͼ����ÿ���ǿ�Ķ��������ļ����ƺ͵�Ѫѹ���ر�����ҩʱ�����˺��á��ĵð�����ǿ�ȵ��ػ�ڷ�����ҩЧӦ������������Ѫ�ǡ��ĵð������ͼ���͵���Ѫ�����ã�����ʱ���ɲ�����Ѫѹ��Ӧ��ʱ�������������غ���ʱ���ܲ�����Ѫѹ�������á�
���ţ�����ͪ���Ұ���ͪ��Amiodarone,Cordarone��
1��ҩ�����á����������ʧ��ҩ������ͪ�ӳ�������λʱ�䣬����������ϵͳ��������·�����������á�һ�ξ���ע�䣨5mg/kg�����������Ҵ������ӳ�A-H���ڣ����ӷ��Ҳ�Ӧ�ڣ�������ӳ��Ḵ��ʱ�䣬�����ӳ�H-V���ں��ķ���Ӧ�ڣ����ڿڷ�����ͪ������ʼ�A-V������Ҳ����ϣ-����ά�������ķ������Ҽ�A-V�Ӧ���ӳ����ĵ�ͼ��������������Ʒ������QT�ӳ���
����ͪ��Ѫ��ƽ�������ɳ����ã��ɽ���״����ΧѪ�����������ӹ�״����Ѫ��������ע�������������������Ѫ�ܣ�ʹѪѹ�½���
��ƷΪ���⻯���0.2g����75mg���ɸ������ڵ��л����״�ٹ��ܡ�
2��ҩ��ѧ�����ղ���ȫ���������ö�20%��40%���ڷ����߷�ѪŨ��Լ4��7h����ע�����ó��ֿ죬15s��51min���������Ϊ15min��3h��2��6h������ʧ����ƷΪ�߶���֬�ԣ����ڷֲ�����Ϊ������ģ�ͣ��ڷ�4��5������ɶ��ļ��ķֲ���dz�ܱ��Ҵ�����ȶ��������ɽ���ѪŨ�ȼ�⡣����������T1/2����15�졢3��������������Ч����ѪŨ��0.5��2��g/ml����ֵ����������Ӧ��ѪŨ����ֱ�ӹ�ϵ����Ҫ��л����Ϊȥ�һ�����ͪ����ѪŨ��Ҳ��ҩ��������仯��С���־��λ�����й��
3����Ӧ֤���������Ժ����Կ���������ʧ������Ч�����Է��������˵�Ԥ��������ת����δ�����߶������������Խ��ͣ����ҽ������۷����Ķ����٣�����WPW�������ٻ����Է�����������Ч��Ҳ�Ϻã������粫��Ч�������Ҳ����������ߡ�
4������֤ ��������ԭ���ض����ͻ�֢״���Ķ�������δװ�����ߣ�����״�ٹ���ʧ���ߣ�������Ѫ��������˥�����á�
5���÷�����������ע��һ����������5mg/kg��ÿ֧150mg��3ml������5%������Һϡ�ͳ�20ml��������ע5min���ϡ��ٴ�ע����15min���ϡ�
�ڷ����������Ÿ�������С����1��ÿ��0.2g��ÿ��3�Σ���2��ÿ��0.2g��ÿ��2�Σ��Ժ�ά����Ϊÿ��0.2g��0.1g��Ҳ�ɳ����ܸ������ϴ�1.2g/d����3�θ�����2�죬�Ժ�1g/d��8�죬0.6g/d��4�죬һ�����ִθ����ٺ�ά����0.2g/d��ÿƬ0.2g��
6��������Ӧ�����̿ڷ�����������Ӧ������Լ30%��50%�����ط�Ӧ�м�״�ٹ��ܿ�������£��輰ʱͣҩ������ά�����ι�����SGPT���ߣ�����Ѫ�ܷ����е�Ѫѹ������ע����죩����˥���շ���QT�ӳ���QTC>0.6s��ͣҩ�������������ͣ�ɫ�س��ţ������У����ģ����أ������ϰ��ȡ�Ӧ���ڲ�ι��ܡ���״�ٹ��ܼ�X����Ƭ��
7��ҩ������á�����ͪ���ߵظ���ѪŨ�ȣ�����ʱӦ���ٵظ�������������ͪ�뻪����ȿڷ�����ҩ����ʱ�����ӳ���Ѫøԭʱ�䣬Ӧ���ٿ���ҩ������
��ʮ��ά�����ף��첫������������Verapamile,Isoptin��
1��ҩ�����á����������ʧ��ҩ��������������ͨ���ƣ����ܻ����ƣ����������봫��������ʱ���ļ�ϸ����Ѫ��ƽ��������ͷ��ҽ�ĵ�ȡ��ͨ����ͨ���ĸ����������ά�����������Կ���������ʧ���dz���Ч��
�������ػ���ά�����鷿����WPW�ۺ������˿����̲�Ӧ�ں��������ҷ�Ӧ�����������Ҳ��������Ӧ������ಡ�ˡ�
����Ѫ�����ã��������������ļ���л��Ҫ���п��Ľ�ʹ���á�
2��ҩ��ѧ���ڷ��������ã���90%�����������öȽ�10%��22%��˵�������ԸΡ�������л���ʶ�����ģ�ͷֲ���T1/2-��Ϊ18��35min������T1/2170��440min��70%������й��16%���ų���90%�뵰��ϡ��ڷ�����������ó�����2h��5h��߷塣��עά�����ף�1��2min�ڳ���ЧӦ��10��15min��߷壬ά��6h��
3����Ӧ֤ ��ע���������������Ķ����٣�������;�����������˰���������ʣ����ڹ�״���������������������ʧ�����ڷ����������ػ����ã����Ʒ������˵Ŀ������ʣ��ʺ����ļ�����������Ĺ��ܲ�ȫ�����������ҽᲡ���߲��á�
4������֤������ۺ�������Ȼ��ȷ������ͣ���Դ���ݿˣ���Ѫѹ�����Ķ��������£����������ص�����˥�ߣ������ػ��ж��������������ڷ��æ����ͼ���˫�����������Ѫƽ���ˣ�Ҳ������WPW�ۺ����鷿�����˲��ˡ�
5���÷�����������ע��0.075mg/kg(����������5.0mg)����5%������20ml��ϡ��ע��2min���ϣ�����Ч��������Ӧ��10min����ظ�1����������������Ч��30min��ɸ�0.15mg/kgһ�������Ϊÿ֧5mg/2ml���ڷ�Ϊÿ��40mg��ÿ6hһ�Σ�2��3�����Ҫʱ��������ÿ��120mg��ÿ6hһ�Ρ�Ƭ��Ϊ40mg/Ƭ��
6��������Ӧ����Ѫѹ����ʱ����10%��������ƻ��Ȼ�����ת���������������Ӱ��������Ķ��������ضȷ��Ҵ������ͣ���ʱ��������������ء�����Ʒ�������ȣ�����˥�ߣ��շ�����أ�����ѣ�Ρ����ġ�Ż�¡����صȡ�
7��ҩ������� ά�����ɼ��ٵظ����������������ѪŨ�ȣ�����ʱ�ظ���Ӧ��������������������ͼ����ã��ر��ڳ�Ѫ���ļ���������˥�ߡ������ļ�����ʱ���Է������ļ����ơ�Ӧ��ά������ǰ48h���Ժ�24h�ڲ�����˫����������á�
��ʮһ������Ʒ��Atropine��
1��ҩ�����á�Ϊ���M-��������Ŀ�����ҩ��ҩ�����ö�档�ܽ������������Ѫ��ƽ�������κʹ̼��йص���Լ�������ƺ��١�֧���ܼ���������ڣ������������������ƣ��������ʣ�ɢ��ͫ�ף�ʹ��ѹ���ߣ��˷ܺ������ࡣ
2��ҩ��ѧ���ڷ��������ã������Ρ�������л������ЧӦ����2h������ȫ��ЧӦ�ɳ���24h��
3����Ӧ֤���������������������������ҩ�������Ķ������������ػ�����ȵȣ��������������İ�-˹�ۺ����������л����ж������������ʹ�ȡ�
4������֤��ǰ���ٷʴ����Ź��衢����ۡ�
5���÷������������ÿ֧Ϊ0.5mg��1mg��2mg���������ಡ���ã�������5mg��10mg�������л����ж��ã���Ƥ�»�עÿ��0.5mg����עÿ��1��2mg��5%������10��20mlϡ�͡��ڷ���Ƭ��ΪÿƬ0.3mg��ÿ��0.3��0.6mg��ÿ6��8hһ�Ρ�
6��������Ӧ���ڸɣ�ѣ�Ρ�Ƥ�����졢ͫ�������������졢�˷ܡ�������ʵȡ�ԭ��ǰ���ٷʴ�ʱ��������������
��ʮ�������������գ������ף�Adenosinetriphosphate,ATP��
1��ҩ�����á�ATPΪһ����Ҫ���м��л����������Ҫ����;��������һϵ���������̣�������״��������ѭ��Ѫ��������ѪС�幦�ܡ�֬����֯�ֽ�ȡ�
ʹA-H�����ӳ����ӻ������ͷ��ҽ��ǰ��������۷���·����ֹ�Ķ����٣����ڴ���ʱ�䡢ϣ��ϵ����ʱ����ޱ仯������н�ǿ�������������ã����ɺ��ͬʱ�������ͣ������������Ķ�������
2��ҩ��ѧ��Ѫ�����Ѹ�٣�Լ30s�����ÿ죬ҩЧ��ʧҲ�졣
3������Ӧ�� �����������Ķ����٣�ά��������Чʱ���ԣ�������ۺ������������ɣ����������ã��й���ʷ����ƷΪ������Ʒ�����á�
���ÿ֧20mg-2ml��ۼ�20mg�������Ỻ��Һ2ml����20mg����������Һϡ�ͳ�5ml��5��20s��ע����Ч�ߣ�5min�������30mg��������������40mg��Ӧ���ĵ�ͼ���⣬�۲�Ѫѹ�仯�Ͳ��˷�Ӧ��
4��������Ӧ����Ʒ�б����������ٸ����ʽϸߣ�88.5%������������Ӧ������Ҳ�ߣ�90.4%�����������ز���ȫ�����ʡ����ơ�ͷ�Ρ�ͷ�͡���֫��ľ����ʹ�����ġ��沿���죻һ��1��2min������л��⣻�ɳ����ͣ��������ȷ��Ҵ������͡������粫�����������Ķ����١�����ȣ�һ���ѪѹӰ�첻��
��ʮ��������Ӣ�ƣ�Phenytoin,Dilantin,DPH��
1��ҩ�����á�����B�����ʧ��ҩ�������ػ��ж�ʱ��ʹ���Ҵ���ʱ��ͷ��ҽӦ�����̣����������ԣ���С�ֿ�Ұ��ά4���¶ȡ�Ƥ�⣬����Ӣ�ƿ��ܽ�������Ѫ����������������ĩѹ����������Ѫѹ�½��������Դ���Ƥ���˶����и߶�ѡ�����������ã�����Χ��ʹ��
2��ҩ��ѧ���ڷ����������������߷�ѪŨ�ȳ�����2��8h�����ҩ�����ó����ڿڷ�7��10���ͣҩ�������Կɳ���1�����ҡ�����ע�����÷�����5��20min������ѪŨ��10��20��g/ml������16��g/ml�����ж������ھ�����������2��4h��ȡѪ��Ũ������ȷ����Ũ���Ƿ��Ѵﵽ���ڷ�ҩ�1�ܺ�ʼ�ɲ��´μ���ǰ�Ĺ�Ũ�ȡ�������ʸߣ�Լ90%��5%��ԭ�������ų�����Ҫ�ڸδ�л����л�����������������ǻ�������������ȩ���ϣ�����֭�����ų��������ȡ���ڸδ�л������T1/2�ڳ���һ��Ϊ24h(20��30h)���β�ʱ���ӳ���
3����Ӧ֤����ƷΪһ�ֶ��߿�����ʧ��ҩ�����ٵ���Ӧ�á�Ŀǰ��Ҫ�������ػ����Է�Ӧ�������λ���ɣ��緿���Ķ����١����������ˡ����硢�����Ķ����ٵȡ�
4������֤ ����ȫ�Դ������͵��������Ķ����٣������ػ����£����Ρ�������˥�ߡ�Ѫ�����ͻ������ã�Ӧ�ʵ�������
5���÷��������������ÿ֧0.1g��0.25g����עÿ��100mg��ע����ˮ5��10mlϡ�ͣ���3��5min����ע�䣬ÿ��5��10min���ظ�����3��4�Ρ�Ϊά�����ƿɸĿڷ���ÿ��0.1g��ÿ��3��4�Ρ�
6��������Ӧ������ע�����������Ѫѹ���ݿˡ��������ơ��Ķ��������������͡����Ҳ�������ͣ�������������ע���ٶ�ÿ���Ӳ�����50mg���Է��䷢����������Ӧ��ͷ�Ρ�ͷʹ������������˶�ʧ������˯�������ϰ������ڷ��ÿɷ������ġ�Ż�¡���ʳ������������Ƥ���ϸ�����١�ȫѪ���ٻ��ϸ����ƶѪ��Ҷ��ȱ���������ȡ�
7��ҩ���������� ���յ������Ƹλ���Թ�������ø��ҩ����ɸı�DPH��л��
�뻪�����˫�㶹�ؿڷ�����ҩ���ã��˴˵�ѪŨ�Ⱦ����ߣ���DPH������ڷ�������ʱ����û������ͪ������˾ƥ�֡��������Ƶ�ȡ��Ѫ������ϣ���һ��������DPH���ã������¡�����ҩ�����������Ƽ������Ƹδ�л��Ҳ����DPHѪŨ�ȣ���ǿ�����ã�DPH�ɼ�ǿ�ĵð����á�
��ʮ�ģ�����������أ���Ϣ�����������Isoproterenol,Isoprenaline,Isuprel��
1��ҩ�����á�Ϊһ��ǿ�ĺϳ��⽻�а��ķ�ѡ���Ԧ����弤��������22-5�����������ʼ��ļ������ԣ�����������ϵĤѪ�ܣ����⣬Ҳ������ΧѪ����������������Ѫ���������������ѹ��������ƽ������ѹ����������֯���˷��Ժʹ�����һ�����ӣ������ļ����������ɳ�ƽ������ʹ��̬��Ӧ�Դ̼�����鰷�ͷż��٣��ɳ������ӹ������������Ҫ��л����Ϊʹ����֬��ϡ�š��ٴ���Ϊ֧�����ż�������̼��������ӷ��Ҵ����Ϳ��ݿ�ҩ�á�
2��ҩ��ѧ���ڷ�������ȫ���������Եġ�������л����С���������á�T1/2Ϊ2.5h��������ҩ�Ĵ�л�����ͬ�ڿڷ������롣����ע���66%��ԭ���ų��������������Ϊ3-��-����л��ڷ����ų���ԭ��ռ6%��10%��3%��11������Ҫ�����3-��-��������ų�������80%Ϊ�������ϵ��������ʽ��3-��-�������Ϊһ�ַdz����Ħ��������������������¸�ҩ����15��30min��ʼ���ã�����45min��2h��
3����Ӧ֤�����ݿˣ��ر�Ⱦ���ݿ˰�����ΧѪ������ʱ����ȷ��Ҵ������ͣ������ڼ����ļ�����ʱ������������
4������֤�������ļ�������Դ���ݿ�ʱ��Ʒ�������ļ����������ã�һ�����Ų��á���Ѫ�����������Ӧ������Ѫ�����������������
5���������÷������ݿˣ��ڳ�������Ѫ��������0.2��0.4mg���������200ml��Һ�����Σ�ÿ֧1mg/2ml�����������ʷ�Ӧ�������ʡ��߶ȷ��Ҵ������ͣ����ʵ���40��/min��0.5��1mg����5%������Һ200��300ml�����Ρ����߿�10mg���º�����ÿ��3�Ρ�ÿƬ10mg��
6��������Ӧ �Ķ����٣�����ʧ�������硢�����Ķ����٣����ļ¡�ͷʹ��ͷ�Ρ��沿���졢����������־�ȡ�Ϊ���ƶ��Ա�Ҫʱ���æ��������ͼ���
7��ҩ������á��������⽻�а�ҩ����������صȣ��������Ⱥ��ÿ��������䶾�ԡ�
��ʮ�壩����
1��ҩ������ ���ڼشּ���ϸ���ڣ���ϸ������Ҫ�����ӣ�����ϸ������ѹ�����ƽ����ڡ��Ǽ������ʺϳɺ�������л���嶯�����Լ��ļ������������ȡ��ļ�ϸ���ڡ����Ũ�ȶ��ļ��������ԡ������Ժ��˷��Զ���Ӱ�졣��Ѫ�������ֿ�Ұ��ά�����ԣ����Ӿ�ֹĤ��λ�������ʣ�ʹ��������ػ�������Ҳ���ӣ����շ���������ʧ������Ѫ�������ļ������ԡ������Ժ��˷��ԡ�
2��ҩ��ѧ������������ʳ����ȡ�أ�ÿ��Լ2��4g�����Գ����ա�����Ѫ��Ũ��3.5��5mmol/L�������Ӻܿ����ϸ���ڣ�ϸ����Һ�й����ļ�����Ѹ�پ��������ų���
3����Ӧ֤�����ػ��ж�����������Ķ����٣��緿���Ķ����ٰ鷿�Ҵ������͡��������Ķ����ٺ�Ƶ�������粫�����ε�Ѫ�أ�����������ά��ƽ�⡣
4������֤������Ѫ�ؼ��ã����������ؼ�������ʱ���ã������ػ��ж��⣬�������������á�
5���÷������� ���ﳣ�õļ����Ƽ��ж����Ȼ��ػ��Ŷ������þ���Ȼ��س���10%�Ȼ�����Һ��ÿ֧1g/10ml������20mmol������һ��ÿ����10%��15%��Һ10ml������5%��10%������Һ500mlϡ�ͣ����Σ���ҺŨ��һ�㲻����0.2%��0.4%��Ϊ��������ʧ����ҪʱŨ�ȿɼ���0.6%��0.7%����10%�Ȼ���10ml(��15%7ml)������������Һ200��300ml�����ĵ�ͼ��»������Σ�����������ע1��2g�ɼ�Ч����ע�������ķ��ʿ�ʼ������Ԥʾ��Ч�����ѽӽ�������ʧ�����ƺ�Ŀڷ��Ƽ���
�Ŷ������þע��Һ��Potassium magnesium aspartateinjection��Ϊ10%��Һ��ÿ֧10ml����2.9mmol������þ1.75mmol���Ŷ��������������ѭ��������ѭ������ϸ������ǿ������Ϊ�����������ʹ�������ط�ϸ���ڣ��൱�Ȼ���1/4�����Ŷ�����أ����ɳ�����������á�10��20ml������5%��10%������Һ250��500ml�������Ρ�
6��������Ӧ�����ι���ʱ�ɳ���ƣ�������������ͣ�������ʧ����Χѭ��˥�ߣ����ʼ�����������ͣ������ҺŨ�ȹ��������ֲ���ʹ�������ס�
��ʮ����þ��
1��ҩ�����á�þ�ͼ�һ��ͬ��ϸ��������Ҫ�������ӡ�����þ47%�ֲ�ϸ���ڣ���0.4%�ֲ���Ѫ�����ڸ����������йصĻ�У�þ��ATP��һ�ָ����ӣ�Ҳ��ϸ��ĤNa+-K+-ATPø�ĸ����ӡ���Ѫ��ʱ��ø���Խ��ͣ�����Ҳ����þ���ط���ϸ����Ҳ�ܵ����ơ�����þע��������������ϵͳ���ɳڹ��������н⾷��������ѹ���ã������ھ��ʡ������֢�����˷缰��Ѫѹ�Բ��ȡ�
ʧ������ҩ����Ӧ�ã����ղ����ۺ��������Ծƾ��ж�������������Ѫ������������þȱ����һ��Ϊ���Ժͷ�չ������
Ѫþ�����½����ٴ��ɼ�����ΪӦ��������֬�����ߵĽ�����糷ͣ�ƾ��������ļ�������������������������¶�ڼ��˺����У�Ӧ�������ټ������ĺ�������ػ��ж�Ҳ��߷����ʵĵ�þѪ֢����������������ʧ�����貹��þ�������ơ�
2��ҩ��ѧ������Ѫ��þŨ��0.75��1.2mmol/L(��1.8��2.9mg/dl)������ʹѪþ��������ʱþ������Ҳ��Ч������þ��ڷ����գ�����������ʧ����Ҫ�þ��λ�ע��
3������Ӧ�á�����þȱ���йصĿ���������ʧ�������Է����Ķ����ٻ������Ķ����١����ػ�����������ʧ�������Ťת�������Ķ����١�����֤Ϊ���¸�Ѫþ�������������ܲ�ȫ�����ഫ�����͡����ػ�������С��Ӧ�á�
����þ����Ϊÿ֧10%10ml��10%20ml��25%10ml����ע��25%��Һ��ÿ��10ml�����Դ�����5%��10%��Һ10ml����5%������Һ10mlϡ�ͺ�����ע��Ҳ�����Ŷ������þ��Һ����ͬʱ�����Ρ�
4��������Ӧ��þΪ���༰���������Ƽ���������ҩ��ע�ɲ���Ѫѹ�½�����������ͣ�����ɲ��������粫���������Ķ����١������ĵ�ͼ���ְ���P-R�ӳ���QRS������Ѫ��þŨ�ȸ���2mmol/L�ɳ��ָ�þѪ֢��֢״���������沿���졢��Ѫѹ�����췴�����ơ��ڻ�����ԡ����½��͡��Ĺ������ơ����������ƣ��������������Ժ������ơ��ɲ�����Ѫ�ơ�
�ж�����Ϊ������ע10%���������10��20ml�����ο�Ѹ�ٶԿ�þ��������ϵͳ�����á�
5��ҩ������á������ػ������ˣ���þ�ж������ʱ�ɲ����������͡��������������ͼ����ã��ɲ������ֵ��������͡�
�塢Ѫ�����ż���������ѹҩ�����Ľ�ʹҩ
��������Ѫ�ܼ���Ӧ���൱�㷺��һ��ҩ��ΪѪ�����ż������������Ƹ�Ѫѹ�����Ժ���������˥�ߡ��Ľ�ʹ����Ĥ�����ȡ�Ѫ�����ż���������Ҫ���ò�λ������Ϊ�����ࣺ��Щ���Ŷ���Ϊ����ֱ���ɳ�Ѫ��ƽ��������Ѫ�ܱڦ����壻��Щ��Ҫ���ž���������Щ�������Ŷ����;�������22-9����
��22-9 ����Ѫ�����ż����ࡪ�����������ò�λ
| ���� | ���� | ����Զ���/���� |
| Ѫ�����ż� | Ѫ�����ż� | Ѫ�����ż� |
| 1��ƽ�����ɳڼ� | �������� | 1�����������ͼ� |
| �±���� | ������� | �������� |
| ��ѹ�� | ��������ɽ�洼�� | ������ |
| 2����2���弤���� | ��������ɽ�洼�� | ����� |
| ɳ������ | �� | 2��ת��ø���Ƽ� |
| ���DZ��İ� | �� | �ϼױ����� |
| �嶡���� | �� | ���������� |
| �� | �� | 3�������� |
| �� | �� | 4�������� |
| �� | �� | ������� |
��Ѫѹ����Ϊ��ΧѪ���������ӣ�ֱ��������Ѫ��ƽ������ҩ��ʹ����Ѫ���ɳڶ�Ѹ�ٽ���Ѫѹ��Ѫ�����ż���������˥�ߵ����û���һ����ͨ�����ž��������Ӿ������������ٻ��ľ���Ѫ�������ͷ�ëϸѪ��ѹ����������ĩ��ѹ��ǰ���ɣ����Ӷ�����γ�Ѫ����һ���棬Ҳͨ������С��������������Ѫ���������Ӷ�����ɣ����������ұ�Ѫ���ܣ������IJ���������Ѫ����ʹ�����������������٣���ëϸѪ��ѹ���ͣ��Ӷ�����˥�ߺ�ת��Ѫ�����ż�����ǰ���ɣ�Ҳ�����ļ������ģ����������ļ�ȱѪ��ͼ22-2����
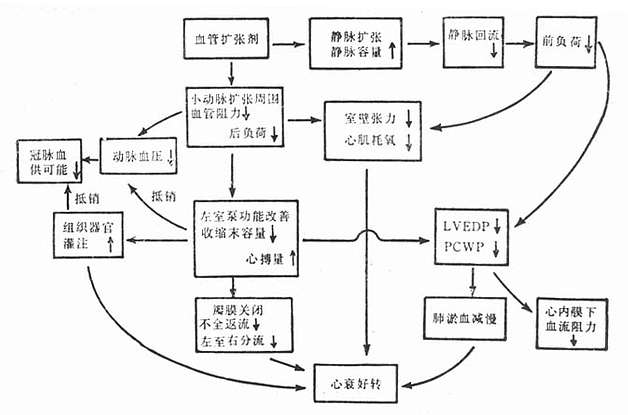
ͼ22-2 Ѫ�����ż�������˥�Ļ���
Ѫ�����ż����շ���Ӿ���������˥���ߵĵ�Ѫѹ����ʹ��Ӧ�������ơ�Ѫ�����ż���ЧӦȡ���������һ��������Ρ�С����ʱ����Ѫ�����ӣ���ëѪ��ѹ���Ͷ�����ѹ���ٱ仯�����ע�ٶȼӿ죬�ڽ�һ����������Ѫ�������ͷ�ëѪ��ѹͬʱ������ѹҲ��ʼ�½������������ٶȣ�������Ѫ������ëѪ��ѹ������ѹ���½�������ʱ������Σ����������˶�����������˥ʱ����С�����Է���������Ч���ͱ���������Ӧ��ʱ������ѭ����ԭ��ֻ���ڰ�γ�Ѫ����ëѪ��ѹ�������ߣ������ߵ���2.0��2.4kPa(15��18mmHg)(�ٴ����ƣ��ε��ކ���)������Ӧ������ҩ���ʹ����ѹ������������Χ�ڣ�һ������ѹ��Ӧ����13.3kPa(100mmHg)��
(һ)�����ƣ�Nitroprusside sodium��
1��ҩ�����á�Ϊһ��ǿ����ЧѪ�����ż���ֱ��ʹС����������ƽ�����ɳڣ�������ΧѪ��������ʹ������Ѫ������Ѫѹ�½��������������ж��Ķ����٣����ļ�����������˥�߿ɸ�������Ѫ����
2��ҩ��ѧ����������ҩ��1��2min����Ч�����ó���5��10min��һ�����������ͳ�5�����軯�����ӣ������ϸ������֯���ϻ���ϣ��γ��軯������ڸ�ת��Ϊ�����Ρ������ư���ڼ��̣�Լ�����ӣ��������ΰ����Ϊ7�죬��ȫ��������й��
3����Ӧ֤ ���ָ�ѪѹΣ����Ѹ�ٽ�ѹ�����Ѫѹ�Բ���������Ѫѹ������������С������������˥�ߺϰ��Ѫѹ����ڳ�Ѫ���ȸ�ϸ�������������в㶯�����������ļ�����ʱ����Դ���ݿ˰�Ѫ������״������˥�ߣ�����ԭ����������˥�ߡ�
4���������÷���������Ϊˮ�ܷۼ���ÿ֧50mg����5%������Һ�ܽ⣨������Һ���ܲ������Բ����Ϊ���ʼѪѹ�½����죬���Ⱦ���5%������Һ�������ٶȣ�8��/min��ʼ�����ٽ������ܺõ�25��50mg����500ml��Һ�С���15��g/min��ʼ�����Ѫѹ�߿���25��g/min��ʼ����ÿ3��5min���Ѫѹ���������٣�ֱ����ëѪ��ШǶѹ����������������Ѫѹ��13.3kPa(100mmHg)���ϡ�����ѪҺ����ѧ��⣬��Ѫѹ�����ߣ��ٶ�Ϊ25��400��g/min��Ѫѹ������Ϊ25��150��g/min���������Ƶ�ͨ����Ӧ��Ѫѹ�����½�30%��40%������˥���ˣ�ע��ʹѪѹ�½�������2.67kPa(20mmHg)���������Ӳ�����20��/min��
������ˮ��Һ���ȶ���Ӧ����ǰ����������Һ����������Ϊ���軯�ƺ��軯�ʹ��Һ��ɫ����֣��ʵ�ע�������õ������ιܾ�Ӧ�ܹ⣨���úڲ�����ֽ����ֽ���ڣ�������ʱ�䳤���ر�������˥����Ӧ���Ѫ������Ũ�ȣ����Կ�ʼ��50��100��g/ml����
5��������Ӧ����ע����ɲ������ġ�Ż�¡����겻����ͷʹ��ͷ�Ρ��عǺ��ʡ���ʹ����������ȣ�Ҳ�ɲ���Ƥ�����졢����֢״����������ֹ��ע��Ѹ�ٺ�ת����ҩʱ�䳤��������������ж�֢״��������鴤�Ծ��Ρ�ȱ���ۺ������������չѸ�ٵļ�״�ٹ��ܼ��ˣ����Է��������ġ���Ѫѹ�����������ɲ�������Ѫ�쵰�ף�ά����B12ȱ�������������ж��������οɱ������������ܰ���Hyhroxycobalamine��Ҳ�����������ж���
�����������������������Phentolamine,Regitine��
1��ҩ�����á�Ϊһ�֦��������ͼ�����ͻ��ǰ���壨��1����ͻ�������壨��2������ǿ���������á����ļ���ֱ�ӵ����Լ����ͱ�ʱ�����ã���Ѫ�ܱ�ƽ��������ֱ���ɳ����ã��ɽ�Ѫѹ����θ���������鰷�����á�
ѪҺ����ѧЧӦ�������ƣ�����С����������Ҳ���;������������Ӿ����������������ߵ���������ĩѹ�����Ͷ���ѹ��ʹ���͵�����Ѫ���������ߡ��������Ķ����٣��������Լ������ã�����������������������5min�����ø߷壬���ó���15��30min��
2������Ӧ�� ��Ӧ֤ΪԤ���������ȸ�ϸ������ѪѹΣ����ǰ��������ʱ��������������ø���Ʋ��˵ĸ�ѪѹΣ���ݿˣ����й���Ѫ������ʱ�����䵱��ëѪ��Шѹ����2.67kPa(20mmHg)�����ľ���ѹ����1.96kPa(20cmH2O)������������˥�ߣ��������ػ��Ͱ�����Ѫ��ҩ©��Ѫ���⣬����Ƥ��������Ԥ���������ȸ�ϸ������ϣ������������飩�������ˡ������߽��á�
���ÿ֧10mg/1ml��10mg����5%������Һ50��100ml����0.1��1mg/min���ʾ��Ρ��ڷ�ÿƬ25mg��
3��������Ӧ��������ҩ����������Ķ����ٻ����������ʧ�����Ľ�ʹ������ͷ�Ρ����ġ��沿���죻�����߿��е�Ѫѹ��������ɲ����Ƕ¡���к��������
4��ҩ������� ����ȥ���������ء���Ͱ��Ⱥ��ã��������ݿˣ������ĵð����ã������ȸ�ϸ������Ѫѹ���˴������������ù���Ӧ�����ػ��Ƽ���
�������������ࣨNitrates�����ﳣ�õ����������(Nitroglycerin,GTN)�����������洼��(����ʹ��Isorbide nitrate,Sorbitrate,Isoket)�����������洼��(Isorbide-5-mononitrate,Elantan)��
1��ҩ�����á����������ɳ�Ѫ�ܱ�ƽ�������ر���ëϸѪ�ܺ��Ѫ�ܣ�������������������Ѫ�أ����ٻ���Ѫ����������������ĩѹ��Ҳ��Ƚ��Ͷ���ѹ�������Ź�״���������������ļ����������������ܴٽ���֧ѭ��������ȱѪ����Ѫ�������Ի����Ľ�ʹ��Ҳ������������˥�ߡ�
�������������ƽ����Ҳ���ɳ����ã��ɻ������ž��Ρ�����ʹ������ʹ�ȡ�
2��ҩ��ѧ��������Ϳڷ�����100%�����δ�л����Ч�����Կ�ǻճĤ��Ƥ�����գ���ҲѸ�ٱ��δ�л��T1/21��2min������������ʾ�����η���Ӧ�ÿ�ʹѭ��ЧӦ���������ԣ�����ɷ���������ͣҩ���ܵ���Ѫ��������
��������ɽ�洼���ڷ���20%��70%���Ρ�������лΪ������֬������Ҳ����Ѫ�����á�T1/2������ʹΪ0.5h��2-��������ɽ�洼��Ϊ1.5h��5-��������ɽ�洼��Ϊ5h��4��6h����5-�������л����������������ڳ����ɲ����Ϻ㶨��ЧӦ��������������͡�������й��Ϊԭ�ε�ҩ�ﲿ�֣�Ϊ���Ĵ�л���
3����Ӧ֤��������ҩ�� ����˥�ߣ����伱���ļ��������£��������ļ������鷢���ص��Ľ�ʹ�������Ľ�ʹ���ڷ����Ľ�ʹҩЧ���ƹ����в����ɿ��Ƶĵ�Ѫѹ��
4������֤ �������ʣ��������������ʷ������Ѫѹ��δ���εĵ�Ѫ�������ɲ������ص�Ѫѹ�����ٹ������㣬����Ѫ˨�γɣ�����ëϸѪ��Шѹ�����͵Ļ��ߣ���ѹ���ߣ��������ˡ��Գ�Ѫ�ȣ�����խ���İ����İ�ѹ����
5���÷����������������Ƭ��ΪÿƬ0.5mg��0.6mg�����º���ע���Ϊ1mg/1m1��50mg/10ml(Tridil)��һ֧��1mg������ǰ��5%��10%������Һ100��200mlϡ�ͺΣ���ʼ����5��10��g/min��ÿ��5min�����Ӽ����������������200��g/min��һ�����0.6��12mg/h����Ч��Ӧ������������Ӧ��ͣ�����Ľ�ʹ������
��Ƥ����Ϊÿ�����������Ϊ0.5��5��10��15mg���ȡ�ÿ24hһ���������ظ���Ƥ����
5-��������ɽ�洼����Elantan��Ƭ��ÿƬ20mg��40mg��ÿ��20mg��40mg��ÿ��3�Ρ�
6��������Ӧ����Ѫѹ�鷴�����Ķ����٣�������ҩʱ���д����ã���Ѫѹ�������ԣ�Ӧ����ֹ��ע��̧��֫�壬�ܿ���ʧ����������Ѫ�쵰��Ѫ֢����������ͼ�������500��g/min��������礣�����Ѫ�쵰��ѪŨ��>1.5%��Ҳ�ɳ�����礡���ҩ�����������礣�Ӧ��������ֹ��ע����Ҫʱ����������2mg/kg��10min�ھ���ע�䡣ͷʹ��������������ļ¡��Ķ����١�ͷ�Ρ����Dz������عǺ��ʡ����ⴤ������ʹ�ȡ�
7��ҩ������á������������Ƶ�������Ѫ��ҩ���ã�������������ͼ�����������á�������ҩʱ��Ӧͣ��������������ҩ�
���ģ��ϼױ����ᣨCaptopril��Capotan��
1��ҩ�����á�Ϊһ��ת��ø��������ø�����Կڷ����Ƽ�������Ѫ�ܽ�����ת��ø���ԣ�����Ѫ�ܽ����آ���ǿ������Ѫ�����ã�������С������Ҳ������Դ�Ի����Ľ��⣻����ȩ��ͪ�γɺͿ������ط��ڣ��Ը�����Ѫѹ�������Խ�ѹ���ã��Ը����غ��������غ�����������Ѫѹ��ѹ�����ԣ��Ե�������Ѫѹ�����������ѹҲ���ԣ���������С������С��������������ǰ���ɣ������Ǻɣ��Ӷ���������ܡ�
2��ҩ��ѧ���ڷ�����Ѹ�٣�1h��ѪŨ�ȸ߷壬һ������70%��ʳ��ɽ���ҩ����������30%��60%��ԭ�������ų�������30%��лΪ����Բ���������30%��T1/2Ϊ4h����������ʱ����ϼ��٣�������ӳ���
3������Ӧ�á����������Ƴ����Ʒ�Ч������ظ�Ѫѹ���������ͣ������淽����Ч�����������������˥�ߡ������������á�
ÿƬΪ25mg��50mg��100mg�����Ƹ�Ѫѹʱÿ��25mg��ÿ��3�Σ���ǰ���ã�����������ÿ��50��100mg��ÿ�������Ϊ450mg����������˥�ߴ�С����6.25��12.5mg��ʼ��ÿ��3�Ρ�Ч����ɼ��������
4��������Ӧ��������Ƥ����ȡ��ؽ�ʹ��ɦ����ζ���ϰ���������֢״��������ҩ����ɷ���������Ĥ����С��������ۺ�������ϸ��ȱ��֢������ϸ�����٣�������ͣҩ����������Ӧ����ʧ����⡣
���壩��ѹຣ��ȼױ������ѹ��Diazoxide,gyperstat��
1��ҩ�����á�Ϊһ�ַ�����������ࡣֱ���ɳڶ���ƽ������������ΧѪ��������ʹѪѹ�����½����������ʣ�������ʹ����Ѫ�����ӡ���ʼ����Ѫ�����½����Ժ����ӡ����������ϸ�������ȵ��أ��ɲ�����ʱ��Ѫ�����ߣ�ͨ��12h��������ʧ���ɲ����ơ�ˮ������
2��ҩ��ѧ���ֲ��ݻ�0.2L/kg���뵰�����Ϊ90%��95%�������ܲ�ȫʱ����ʼ��١���Ʒ��ͨ��̥�̡�T1/2Ϊ20��40h(���ˣ���55%��60%����л���������ų�������ע�䣬Ѫѹ�½���1��5min�����������10��20minѪѹ���Ѹ�ٻ�������Ѫѹ���ó���2��12h���ڷ�100%���ա�
3������Ӧ�á����ڸ�ѪѹΣ��Ѫѹ�Բ���������Ѫѹ�����������Ϊ��Ѫ��ҩ�������ȵ�ϸ��������Ԫ�ۻ�֢�����ط��Ե�Ѫ��֢����ĵ�Ѫ�ǡ�
�������ȸ�ϸ�������뵥������ø���Ƽ���Ӧ�йصĸ�ѪѹΣ�̷�����������խ������������ĸ�Ѫѹ���ϲ�����˥�ߡ�ȱѪ�����ಡ�����ĸ�Ѫѹ���������ҩ������ߡ���������ܲ�ȫ���á�
���ÿ֧300mg����ר���ܼ�20ml�����Ƹ�ѪѹΣ��ע������75��150mg(1��3mg/kg,�����150mg)����λ���پ�ע��С���룬�ı�����Ѫ������ϣ���Ҫʱ5min���ظ�1�μ�����5mg/kg��
4��������Ӧ ���������ز�����ӦΪ���ס���˥���أ��ظ���ҩ���У�����Ѫ�ǡ������������ز�����ӦΪ��Ѫѹ���Ķ����٣��Ľ�ʹ���ļ����������ص����ߡ��ɳ���һʱ����ȱѪ�����ȸУ�ͷʹ�����ġ�ʧ�ߡ����ء��������ʵȡ��������ص�Ѫѹ��ʱ���ö�Ͱ����Ķ��������ĵð���
5��ҩ
���۴˴�ȱ��һЩ���ݣݡ�
�ʼ�������?.25mg��1��2h��������Ѫѹ�½���������
4��������Ӧ��һ�����������������������ϴ������>0.5mg/d����������˯���������ӡ���ή����к�ȣ�����������ɭ�ۺ�������������֢�������Ѫ��������Ӧ�ÿ�����̥������ϵͳ�ϲ�֢��
5��ҩ������� �����ػ����ã����������ػ����ԣ����ػ�Ҳ������Ѫƽ���������ã��ɷ������Կ���������ʧ�����Ķ�����������ᶡ���ÿ������ļ����ƺͽ�ѹ���á����������������ѹҩ���ã�����ǿ��ѹЧӦ����Ѫƽ��ѹ���ÿɱ��⽻�а��ࣨ����Ƽ��������������������������़����併ѹ���úͲ����Ķ�������������������ҩ������ǿ��Ѫƽ��ѹ���ã�����ǰ����Ӧͣ��Ʒ�����յ�����ǰԤ���ð���Ʒ��ֹ�����Ķ��������뵥������ø���Ƽ����ÿ�ͻȻѪѹ������������֢��
��������ҩ����˨ҩ
��һ������
1��ҩ�����á���һ��ճ��������������ṹ�������ǰ�����ǿ����ɣ��ǿ���Ѫ���Ե���Ч���š������ж������á�
��1������Ѫ������Ѫ������Ӧ�Ķ�����ڣ�ѪС��ճ�����ۼ����Լ���ά�����ܽ�ϵͳ����Ӱ�졣���صĿ����������������뵰���ʣ�������Ѫ��������ö����𡣸����迹��Ѫø��Ϊ�����ӣ���Ѫø����Ѫ���Ӣ����������ȣ����Ǿ�����˿����Ϊ�������ĵĵ���ˮ��ø�������뿹��Ѫø���������л���ϣ������ռ乹��ı䣬ʹ�����������˿���Ჿλ��Ӧ���������������Ѫ���ӵĴ������ԣ���Ҫ������Ѫø�ͼ������Ѫ���Ӣ�����������������Ѫ������ռ����λ�õļ������Ѫ���Ӣ���ʹ��Ѫøԭ����תΪ��Ѫø������Ҳ�������γɵ���Ѫø���ԣ�ʹ��ά����ԭ����ת��Ϊ��ά���ף�ʹ��Ѫʱ���ӳ�������Ҳ������Ѫø�Ԣ������������ӵļ��ѪС���3�����ͷŵȷ�������á�����Ҳ������ά�������µ�ѪС��ۼ���
��2������֬����֬��ø����ʹѪ֬����ת�ˣ�֬Ѫ���塣
2��ҩ��ѧ���ڷ���Ч������ע�䡣���β��ֱ�ѪС���4�����к͡���Ҫ�ڸδ�л��80%�����ݿ�ʱ��Ѫ�������٣���л������20%��ԭ�����ų���T1/2�����ڼ�����С����ʱ��<5000IU�����Σ�ԼΪ1h���ϴ����ʱ2��6h����ͨ��̥�̣�������֭��й�����������
3������Ӧ�á����ؿ�������Ѹ�٣�����ڶ̣������ڶ������迹����ڷ�������֮ǰ��
��Ӧ֤Ϊ���ƺ�Ԥ��Ѫ˨�γɺ�˨���������ز��ȶ����Ľ�ʹ��Ԥ���ļ�����������ף���˨����֫�嶯��˨���ȣ�������Ѫ������Ѫ�����������������ѭ��������֤�г�Ѫ���ʻ���Ѫ�ϰ����ߣ���������´���δ���ߣ���������������ߣ��ݿˣ���Ⱦ������Ĥ�ף����ظΡ������ܲ�ȫ�����Ը�Ѫѹ���ߡ����������ã�ѪС�����Լ��ٻ���Ӧ���������ж���Ӧ�Ⱦ������ж�����˾ƥ�֡�����ø�ȱ�������غ��á�
�������ÿ֧12500IU��1ml�൱100mg����������Ϊ1mg/kg(��5000IU)����ע�����5%��10%������Һ��������ˮ100ml��ע��ÿ����20��30�Ρ�ά������1000��1500IU/h������ÿ6h���Σ��������Һ�ã�0.5mg/kg��Ԥ���õͼ�������5000IU�Ƥ�£����ڻ����գ�ע�䣬ÿ8��12hһ�Ρ����˼�ע��������ע�䲿λѪ�ס���������ݲ��顢���ڡ�����Ѫ֢״��ʵ���Ҽ����ʱ�����������������Ӧ���廯��
��Ѫʱ�䣨�Թܷ���Ϊʵ�ü�ⷽ�����ɴ��Խ��С���ҩǰ�⣬�´θ�ҩǰ1h�ظ���ʹ��Ѫʱ�����������ֵ��2��3����20��30min��������ֵΪ8��12min���������Ѫ��øʱ�䣨APTT��������ֵԼ35s����Ϊ����ָ���˿���������ֵ��1.5��2.5������60��80s������Ѫʱ�����30min����APTT����100s���ʾ�ѹ������Ŀڷ�����ҩʱ��Ӧ��3��5���ص���
4��������Ӧ���Է��Գ�Ѫ��Ϊ��Ҫ�IJ���֢���������������ڽ���֤���˼���Ա��⡣ѪС����٣������ԣ���ż�й�����Ӧ����ݡ������ס����Ĥ�ס����ȵȡ�����ʹ�û��ߣ��ɷ�����ʱ��ͺ�����������ɵȡ�
�����������Ѫ֢״������ڽ��ɣ���Ѹ����ֹ������ʱ�������㾫���ף�Protaminesulfate���кͣ�1%�㾫����5ml���к���50mg(������ע)�������һ�θ���ע����2h���㾫���������ɼ��롣��������������Ѫ������Ѹ���к��أ��㾫��������Ϊ���ص�1.5��2����������㾫���ױ����������Ѫ��������Ӧ�ȣ�����Σ��������
5��ҩ������á���˾ƥ�ּ�����ˮ�����Ƽ������ø������ƵIJ��ˣ����������Ѫ���ڷ�����ҩ������Ӹ������á����鰷ҩ�����ػ������ᶡ����ù�ء��Ļ��ص����Ƹ��صĿ������á�
��Ѫ˨������ָ��ҩ��������������ά�����ܽ�ϵͳ��ʹѪ����ԭת��ɴ���Ѫ���أ�����������ά�����⣬�Ӷ��������γɵ�Ѫ˨�ܽ⡣��Ѫ˨���ƿ�ʹѪ��Ѹ�١���ȫ���ܽ⣬����Ѫ����֢������Ҳ�ϸߡ�Ŀǰ�����ٴ�����Ҫ������ø����ø�������о��е���֯��Ѫ����ԭ�������TPA������Ϊ��ǰ;��ҩ�
����������ø��Streptokinase,SK��
1��ҩ�����á�Ϊһ�ּ�ӵ�Ѫ����ԭ��������ɦ�-��Ѫ��������������ķ�ø�����ʣ���������Ѫ����ԭ��Ѫ�����γɸ��������л��ԡ�ѭ������������Ѫ˨��������������������Ѫ˨�е�Ѫ����ԭ������Ѫ˨�������ܽ⣻ѭ������������Ѫ���غ�Ѫ���еĿ�Ѫ���ؽ�ϣ���Ѫ˨�γɲ�λ�ͷţ�����Ѫ˨����Դ���ܽ⡣SK��һ�����嵰�ף������������߷�Ӧ������ҩ��ЧӦ�������������Ӧ�����������������Ⱦ�Ļ��ߣ����ڼ��������ڿ��������ø���壬���״μ������볬����˨����Ҫ�����к����ֿ��塣
2��ҩ��ѧ��T1/2Ϊ10��12min��
3����Ӧ֤����˨���������Ѫ˨�γɣ���Χ����˨���������ļ�������֢״��ʼ6h�ڣ���������Ѫ˨�γɣ�Ѫ�����ˣ���
4������֤�����Խ���֤Ϊ����ڳ�Ѫ���½���������Ѫ�����⡢��������2�����ڣ����жȵ���Ѫ�����ϰ���ѪС��ȱ�ݻ��Ѫ���ʡ���Խ���֤Ϊ��ƴ����������ˣ������Լ���δ��10�죻δ���Ƶ����ظ�Ѫѹ���ķ����������ܴٷ���˨��������������δ��10�죻�ġ��θ��պ���߹ǹ��ۣ�ԭ���ڿƳ�Ѫ�����ߣ���֧�������ţ����صĸΡ������ܲ�ȫ��ŧ����Ѫ˨�����ס�
5���÷�����������ʼ����ǰӦ�ⶨ��Ѫʱ�䡢��Ѫøʱ�䡢�������Ѫ��øʱ�䡢��Ѫøԭʱ�䡢Ѫ�쵰�ס�ѪС�������Ѫ���ѹ���Գ�����Ѫ�ϰ�����Ϊ�Ժ���Ļ���
�ۼ�ÿƿ100000��600000IU������ʱ150��IU��60min�ڳ�����ע���������ָ���Ե���������ʹ��Ѫøʱ���ӳ���������2��4������ʼ����ǰ�����ø��ػ�ڷ�����ҩ�����APTT����Ѫøԭʱ��ָ����ٿ�ʼ��ҩ���ƣ��ñ�ҩ�������ø���������Ԥ��Ѫ˨�γ��ٷ���
6��������Ӧ
��1����Ѫ��Ϊ����IJ���֢������������ֹ���ƣ�������Լ25%���������پ������̴��������⼡��ע�����ȡ�걾����ͬʱʹ�÷���̴�����ҩ��ڷ�����ҩ���Լ��ٳ�Ѫ�ķ�������Ѫ������һ����ֹ��ע���ܽ���Լ�����ֹ�����Ѫ���أ�Ӧ����Ѫ�������ʱ���Ѫ������ʱ���ð����ᣨEACA���ױ��ᣨPAMBA������Ѫ����ԭ���羲�����̲�λ������Ѫ���ý���EACA��ֹѪ�ް�ѹֹѪ��
��2��������Ӧ����SK�����߷�����Լ15%������ݡ������졢ͷʹ��ż����֧���ܾ��Ρ���Ѫѹ�������⻯�����ɾ���ע�䣨100mg����ΪԤ���䷢�����ɸ�ҩǰ30min��ע���Ǹ�25mg����ע��������2.5��5mg��
��3�����ȡ�������Լ30%��
���������أ�Urokinase,UK��
1��ҩ�����á���һ������ϸ���ϳɵĵ���ˮ��ø��ΪѪ����ԭ��ֱ�Ӽ������������֯�����к��д�����������ǰ����ʽ���ڵ���ø��ͨ������ˮ�����ö��γɾ�������ø���ͷ���ϵ�п���Ϊ�ٴ���˨ҩ��������ȡ�ĸߡ��ͷ���ϵ�п���Ϊ��˨ҩ����øֱ��ʹѪ����ԭת��ΪѪ���أ�����ά����ԭ������Ѫ���������ܽ����á�
2��ҩ��ѧ��T1/2Ϊ12��16min.
3������Ӧ�á���Ӧ֤������֤��ͬSK�������ÿƿ�ֱ�Ϊ6000��60000��250000IU�ȡ�������Ϊ40��IU/min������ע�䣨10min�����Ժ���40��60��IU��1��2h�ھ��Ρ�UKĿǰ�۸�
4����������Ӧ��Ҳ�������Ѫ�����ȣ��������������Ӧ���˵㲻ͬ��SK��
���ģ���֯Ѫ����ԭ�������Tissueplasminogen actirator,t-PA��
Ϊһ��Ѫ����ԭ��ֱ�Ӽ����������ά����ϵ�������ǿ��������ء�t-PA��Ѫ˨�ڵ���ά����ϳɸ����壬������Ѫ����ԭת��ΪѪ���ض��ܽ����ʵ���ά���ף�t-PAֻ����ֲ������˶�������ȫ����˨״̬����Ϊǰ��������������t-PA��Ҫ����Ѫ�ܱڣ�����Ƥϸ���ϳɣ��������ͷš�1983�����˺�ɫ��ϸ����ϸ�������ҳ�Я��t-PA�Ļ���ͨ��DNA���鼼�����ڴ˾��н��б�����ܴ�������t-PA��t-PA���߿�ԭ�ԣ��������������Ӧ���������ڶ��ݣ���������˨ʧ����ʱ���й�״������·���ź�״�����γ�����Ϊ����ǰ;����˨ҩ�����Ӧ�õ���Ӧ֤ͬSK������10mg��10min�ھ�ע�����Ե�1h����50mg,��2h40mg��
�����
�����
[1] Pentel P,et al: Drug therapyfor cardiac emergencies:pharmacokinetic consideration.In: scheinman MM(ed):Cardiac Emergencies.p288,Saunders. philadelphia. 1984
[2] Goldberger E(ed):Teatment ofCardiac Emergencies 4th ed.p323,Mosby,St.Louis. 1985
[3] Leier��CV(ed):CardiotonicDrugs.A clinical Survey pp85,199,Marcell Dekker.New York. 1982
[4] Ewy GA,Bressler R(eds):Cardiovascular Drugs And the Management of HeartDisease.p57,69,95,103,157.Raven. New York.1982
[5] Conh CR(ed): Cardiac DrugTherapy.Davis.philadelphia.1984
[6] Gould LA(ed:) Drug Treatmentof Cardiac Arrhythmias.Futura.Mount Kisco.1983
[7] ����ǫ�ȣ����ࣩ���±�ҩ��ѧ����12�档�������������磬1985
[8] Bochner F et al(eds):Handbook of Clinical Pharmacology.2nd ed .Little Brown.Boston.1983
[9] British National FormularyNo.9 London.1985
[10] Gillies Hc,et al(eds):ATexbook of Clinical pharmacology.2nd ed. Hodder and Stoughton.London 1986
[11] Gilman AG,Goodman LS,etal(eds): The pharmacological Basis of the Therapeutics.7th��edp716,1338.MacMillan.New York ,1985
[12] Laffel GL et al:Thrombolytic Therapy :��a new��strategyfor the treatment of��acute��myocardial infarction.N Eng1 J Med 1984; 311:710,770