第4章 肾脏微循环及其内在的调节
近年来,由于科学的发展和先进技术的应用,人们对于肾脏微循环及其自身的调节机制,有不少新的看法和研究,也为急性肾功能衰竭、急性肾缺血等临床问题提供了新的理论基础。
第一节 肾脏微循环结构
了解肾脏微循环的生理意义,首先必须了解肾脏微循环的具体结构。众所周知,肾脏可分为皮质和髓质,大部分肾小球分布在髓质,另一部分则分布在皮质接近髓质部分,称之为髓旁肾小球。所有肾单位,包括肾小球、肾小管、集合管的结构都相互一致。但和以往看法不同,目前认为按肾小球所处的部位不同,其毛细血管和小管亨利襻的结构并不完全相同。从肾小球而言,髓旁小球比皮质浅表部位的小球体积大,而且在小球入球动脉和出球动脉之间,有血管联接或称旁路联接,这点在皮质浅表部位的肾小球很少见。髓旁或皮质深部的肾小球不仅体积较大,其单个肾小球滤过率也比浅表部位肾小球高30%~40%。
全肾血流首先通过肾小球,经出球动脉,然后分支灌注肾小管。和过去了解不同,目前认为每个肾单位的小管周围毛细血管并非只限于来自其本身肾小球出球动脉,而是每个肾小球出球动脉同时供应几个肾单位的小管血流。换而言之,每一支肾小管常同时接受几个肾小球的出球动脉血流供应,形成肾小球出球动脉和小管之间错综复杂的交叉。
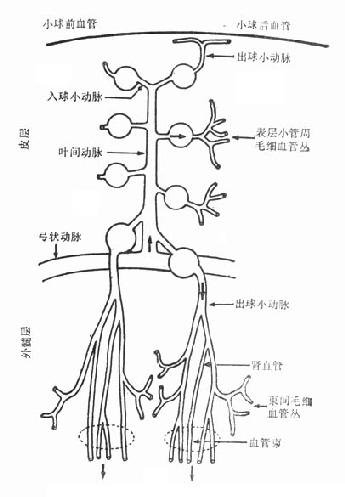
图4-1 肾脏微循环结构
皮质肾小球和髓旁肾小球的血管形态不一致,其生理意义也有区别。球前血管,如入球动脉,从其主支分出来时,有不同角度之分。皮质中层水平的肾小球,其入球动脉较短,按直角方向从小叶间动脉分出;皮质浅表部位肾小球入球动脉处在小叶间动脉末端,形成后者的直接延伸;髓旁肾小球入球动脉和小叶间动脉之间则呈直角或锐角。动物微灌注实验,可见浅表肾小球对一些物质积聚较多,血液到达该部位后,受上述影响,红细胞含量也增加(图4-1)。
肾脏小叶间动脉自主支弓形动脉分出以后,逐渐走向皮质浅表部位,其血管内压力从根部至末端逐渐减弱。按理肾小球处在小叶间动脉根部和其尾部者,其所接受的压力不一样,肾小球内毛细血管的压力应有区别。但实际测定结果,浅表肾小球和深层肾小球内毛细血管压力并无差异。说明深层肾小球入球动脉对球前血管压力有平衡作用,即其血管阻力应比浅层小球入球动脉的阻力高,才能维持这种肾小球内压力的一致性。临床上,慢性高血压引起肾小球硬化,首先出现在深层肾小球,可能和上述解剖部位所受压力强弱不等有关。
关于肾小球出球动脉目前的认识也有进展。肾小球位于浅表皮质部分者,其出球动脉很少分支,向外延伸,主要供皮质部分肾小管的血运。皮质中层肾小球的出球动脉很短,分支构成毛细血管网,围绕肾小管,而且不走向肾髓质。髓旁小球则不然,其出球动脉除少数分支在皮质,大部分分支很长,直接延伸下降到髓质,第一支出球动脉在髓质中分成约30条直小血管降支,呈束状下行,并依髓质不同的层次,形成分支,构成毛细血管网围绕小管,毛细血管网最后汇集成直小血管升支,引流髓质的静脉血。
就大多数肾小球言,出球动脉比入球动脉管径小,肌层较薄。然而髓旁肾小球则有较宽管腔和相对较厚的管壁,如前所述,此结构可能代偿调节血管阻力。实验证明,容积不足或容积扩张,常影响浅表肾小球滤过系数,但对髓旁肾小球影响较小。提示肾小球血流滤过率的调节在皮质浅层和深层的肾小球并不完全相同。
肾小球内毛细血管介于入球动脉和出球动脉之间。出球动脉的阻力变化和阻力调节对维持肾小球内毛细血管阻力很重要,同时出球动脉对其下行的毛细血管和肾小管的回吸收有重要影响,而且毛细血管内压力应保持一定的低水平。鉴于出球动脉的长短、肌层的厚薄,在肾脏各部分肾小球之间并不一致,肾小球毛细血管襻和肾小管周围毛细血管之间的压力差,可能亦不相同。
入球动脉进入肾小球囊后,即形成毛细血管襻,并分成几叶。在较大的毛细血管之间,有较小的毛细血管分支,使小叶之间发生联系。毛细血管最后汇集一处,结合成出球动脉。毛细血管壁分为内皮细胞、基底膜和上皮细胞。系膜细胞存在于小球毛细血管之间。内皮细胞之间具有小窗样孔道,直径为500~1000
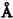
肾髓质血流仅占肾脏血流量的小部分,但髓质须浓缩大量血浆,最后形成每日1500ml左右的尿液排出。
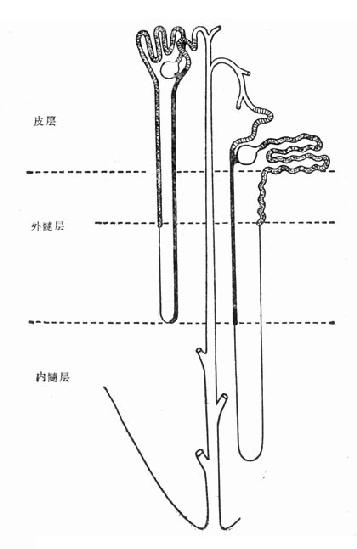
图4-2 内、外髓层毛细血管形态
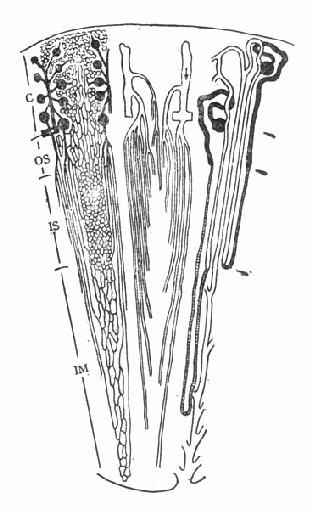
图4-3 叶间弓形静脉
髓质微循环从髓旁肾小球出球动脉以后开始,髓质部分可分为外髓层(outer medulla)和内髓层(inner medulla),外髓层又分为外带(outer stripe)和内带(inner stripe)。外髓层包括小管升降支、细降支和粗升支以及集合管,血管束和毛细血管网也交织在该部分。内髓层包括亨利升降支和集合管,毛细血管呈直血管型态(图4-2)。如前所述,出球动脉,进入髓质外带后,形成马尾状20~30分支,称直小血管降支,呈束状走向,向下延伸,形成毛细血管丛,在内带区比较稠密,到达内髓层,则比较稀疏。最后与直小血管升支汇合,引流入叶间或弓形静脉(图4-3)。
第二节 肾脏微循环的滤过和回吸收
肾小球滤过和小管周围毛细血管的液体交换和回吸收都受相同原理所支配,即各种不同的力和其相互之间的作用可以用Starling滤过和回吸收原理来解释。液体(Jv)通过细胞膜程度是由几方面相互作用而定,包括静水压、胶体渗透压和膜的滤过分数,即:Jv=kf(△p-σ△π)。
Kf为滤过系数,△P为毛细血管内静水压,△π为毛细血管内胶体渗透压。σ为渗透压反应系数,一般在0与1之间,当σ等于1时,胶体渗透压达到理论上最大值,当σ小于1,则胶体渗透压仅有部分作用。肾小球和小管周围毛细血管系统,其σ均接近于1。就整体肾皮质言,σ可以考虑为一个一致的数字。由于正常肾小球毛细血管对大分子物质滤过有高度限制作用,中分子物质如白蛋白亦极少滤过。因此,近端小管内蛋白量很少,肾小球包曼囊中蛋白浓度亦很低,可以不计。肾小球滤过率(GFR)可以公式表示:
GFR=kf(g)(Pg-Pb-πg)
Pg为肾小球平均静水压,Pb为包曼囊中静水压,πg为肾小球毛细血管内胶体渗透压。
肾小管周围的毛细血管的液体交换,可用小管周围毛细血管吸收(peritubular capillary uptake PCU)来表示,即:
PCU=kf(p)[Pc-Pi-(πc-πi)]
Pc代表小管周围毛细血管内压力,Pi代表间质液体压力,πc代表肾小管周围毛细血管内胶体渗透压,πi代表间质液体平均胶体渗透压。
肾脏动脉系统对于血流产生很小阻力,随血管向前走向,其静水压逐渐降低,在入球动脉水平部位下降明显。根据动物(狗)试验,入球动脉压力为6.65~8kPa(50~60mmHg)。有人认为,皮层浅表的小动脉不具备自动调节,并且随动脉压力改变而改变。肾脏的自动调节,主要在入球动脉水平(下文将述及)。正常生理情况的维持,须保持小球内一定水平的静水压(Pg),因为该压力是滤过的主要正压,和胶体渗透压作用方向相反。当肾小球毛细血管内血液流动和滤过后,血浆蛋白浓度逐渐升高,其胶体渗透压也逐渐升高,静水压和胶体渗透压的压力缩小,滤过也减少。随着肾小球滤过过程,毛细血管内血球比积也升高,并经出球动脉流出肾小球,分布至皮质小管周围毛细血管或髓质毛细血管网。由于出球动脉的阻力,毛细血管内的静水压下降,当达到小管周围毛细血管时,已呈相当低的水平。这一点很重要,因为只有使小管周围的毛细血管压低于胶体渗透压,才可以保证回吸收。当回吸收到一定程度,血浆蛋白逐渐稀释,回吸收力也渐下降以至消失。
第三节 肾脏微循环的生理调节
一、内在的自动调节
现已公认,大部分人体微循环床具有内在的自动调节机制,肾脏亦不例外。肾脏微循环具有高度的自动调节来维持总的肾血流(RBF)和肾小球滤过率(GFR)的稳定。当身体动脉压AP高于平均压10.64kPa(80mmHg)时,RBF和GFR均保持恒定。除动脉压改变外,静脉压上升,输尿管压力变化,以及血浆胶体渗透压改变,肾脏血管阻力发生适应性调节。这是一种负反馈控制系统。这种调节即使在去神经的游离肾上仍然存在,具有内在自动的特点。
据实验,当灌注压从16kPa(12mmHg)降至10.64kPa(80mmHg)时,肾小球毛细血管血流率(GBF)仅少许下降,肾小球毛细血管静水压(PGC)亦仅少许改变,即从16kPa(45mmHg)降至5.33kPa(40mmHg),当灌注压进一步从10.64kPa(80mmHg)降至8kPa(60mmHg),PGC从5.33kPa(40mmHg)下降至4.64kPa(35mmHg),GBF明显下降。GBF之所以能在16~10.64kPa(120~80mmHg)时自动调节,主要由于入球动脉阻力(RA)改变。动物(鼠)试验证明,原发性高血压收缩压在21.33kPa(160mmHg)一组和14.63kPa(110mmHg)一组比较,PGC完全相同,两组GBF也相等,但RA在高血压一组比正常血压组明显升高。
入球动脉是自动调节产生阻力变化的主要部分,在正常情况下,对动脉压改变,产生相应的血管阻力变化,而肾小球后的出球动脉阻力改变很小。但当肾动脉压维持在低水平,肾内血管紧张素Ⅱ产生,也可以使球后阻力增加。因此,可以认为肾脏微循环各部位都有自动调节能力,但反应程度不一致。动物试验也证实这点,如动脉压很高时,全部肾单位都有自动调节,但皮质深部肾单位具有更大的自动调节。当血压很低时,深部肾小球血流比浅表小球血流有较大保留能力。这种皮质浅表小球自动调节能力较小的现象,可能和该部位动脉离主干较远较长有关。
关于肾脏自动调节机制,尚未确切了解,目前大多数研究支持两种学说。
(一)肌原学说 小动脉平滑肌的收缩和舒张,可以调节血管阻力,平滑肌细胞对血管张力的增加或减少很敏感,当灌注压增加,首先使血管壁扩张,随之引起血管收缩反应,因而导致血流回复至控制水平。按Laplace公式,T=P×R,T代表血管张力,P代表压力,R代表血管的内径。当灌注压增加,则P值上升,引起R增加,结果使血管张力上升。继以血管收缩反应,调节T至原有水平,使血流保持相对稳定。
(二)致密斑反馈学说 肾小球在入球动脉和出球动脉进出部位有一三角区,即肾小球旁器部位。肾小球旁器包括远曲小管在穿过皮质时,与入球动脉靠近,后者内皮细胞和肾小管上皮细胞在相接触部位,形成肾小球旁器特殊结构。其中有三种细胞:①颗粒细胞,为合成与贮存以及释放肾素的场所;②致密斑细胞,生理上对来自小管液体内所残存的钠和渗透压高度敏感;③系膜细胞。
流经致密斑细胞的小管液体中所含钠,可能通过系膜细胞而刺激颗粒细胞,使之释放不同程度肾素,进入邻近的入球小动脉后,与血管紧张素原结合而形成血管紧张素Ⅰ,并转变为血管紧张素Ⅱ。后者为高效的收缩血管物质,作用于邻近入球动脉,从而影响和调节GFR。
致密斑反馈学说将肾小管功能和肾小动脉对GFR的调节看作一个整体,解释了很多问题,对认识肾脏自动调节机制有重要贡献。但目前仍有争议,争议的焦点在于该学说认为肾小管流量和流经致密斑钠的含量增多量刺激肾小球旁器产生肾素的条件。即远端小管流量增加导致致密斑NaCl浓度上升,引起肾小球旁器肾素加多,从而使局部血管紧张素Ⅱ亦加多,加大了入球动脉阻力。
但根据动物实验,并非普遍如此,有时甚至出现相反现象,即肾素释放的增加,不伴有远端小管钠的增加,甚至是减少。因此,此一学说仍须进一步研究。
二、肾素血管紧张素对于肾内血液动力学的调节
(一)肾素血管紧张素系统成分的肾内定位 如前提及,肾素由肾小球旁器中细胞形成。业已证明,内源性肾素分泌和肾小球旁器颗粒细胞出现颗粒程度有密切关系,即高度肾素分泌时,颗粒化程度也高,反之,颗粒化减低,甚至呈无颗粒状态。目前已知一些因素可以引起颗粒化增高和肾素分泌增多,如肾缺血、长时间缺氧、低钠血、妊娠和肾上腺功能不全等。另一方面,钠负荷、动脉血压升高、体液过多,均可使颗粒减少而肾素分泌下降。调节肾素分泌的机制,可以归纳为几方面:①肾内者,包括肾血管感受器,如肾小球细胞本身和致密斑。②交感神经系统,包括肾神经、循环中儿茶酚胺。③体液因子,循环或局部产生者,包括加压素、血管紧张素Ⅱ和电解质,这些直接作用或间接通过其他调节机制作用于肾小球旁器细胞。
以往认为肾素分泌后,直接进入血管。现在认为进入血管中的肾素,不可能很快到达肾间质,而且肾脏淋巴组织和间质中的血管紧张素Ⅱ和肾素浓度相对地比较高。因此,可能肾素形成后主要是进入肾脏间质的间隙,在该处作用于底物,形成组织内血管紧张素Ⅰ,继而肾素和血管紧张素进入血循环。这一过程主要发生于小管周围毛细血管内。应用免疫化学技术,现已进一步了解肾素-血管紧张素系统在肾内的分布,如图4-4所示。
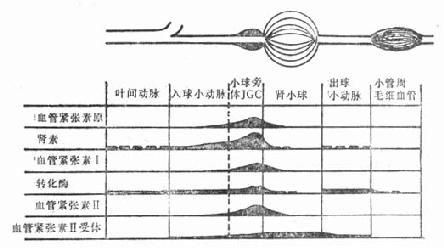
图4-4 肾素-血管紧张素系统在肾内的分布
(二)血管紧张素的肾内血管作用部位 很多研究,证实一些组织的血管平滑肌细胞上有血管紧张素受体,同样也证实肾脏血管平滑肌细胞对血管紧张素有高反应。但肾脏大的动脉例外,不具备这种明确反应。血管紧张素具有三种人们所熟知的生理效应,即:①引起小动脉收缩。②对肾脏有直接作用,小剂量可以引起钠潴留,大剂量引起尿钠增多。③作用于肾上腺皮质,引起醛固酮分泌增加。
近年来,对其肾内的作用研究较多,还证明肾小球内有血管紧张素受体,特别是肾小球系膜有血管紧张素依赖的收缩性,即肾小球结合血管紧张素部位,主要在小球系膜细胞。当血管紧张素Ⅱ加入系膜细胞,则系膜细胞呈现类收缩现象,但肾小球上皮细胞不具备此一性质。对于肾小球细胞收缩能力研究,可以认为血管活性物质如血管紧张素Ⅱ直接刺激系膜细胞,导致了肾小球体积缩小,减少毛细血管通过和滤过面积,从而调节肾小球滤过率,这些研究,提示在血管紧张素作用下,系膜细胞系肾小球毛细血管和液体通过的主要调节环节。
(三)血管紧张素肾内直接作用对血液动力学影响 血管紧张素Ⅱ引起RBF降低,但对GFR较少作用。通过微穿刺研究,当血管紧张素Ⅱ输注时,引起SNGFR下降,小球血流减少,肾小球出入动脉阻力增加,小球滤过常数kf降低。微穿刺还发现,当血管紧张素Ⅱ直接输入小管周围毛细血管,则其阻力增加,因此血管紧张素Ⅱ也影响小管周围毛细血管动力学。有人试验,在同一个肾脏上,两支不同动脉,在其中一支注入血管紧张素Ⅰ,则明显出现血流减少,而未注入者则否。因此,可以认为肾血管对直接输入血管紧张素Ⅱ的前体Ⅰ,可以很快在局部形成血管紧张素Ⅱ,并且引起肾脏血液动力学改变。
三、前列腺素PG(Prostaglandin)
PG由体内多处组织所合成,肾脏是其中重要器官之一,肾脏集合管、髓质、小球以及肾血管细胞都能形成PG,其中主要者为PGE2和PGI2。PGE2可以在髓质和皮质中形成,而PGI2主要形成于肾皮质。
PG对肾脏血液动力学影响,主要是通过动物试验来证实。已经证明PGE2可以增加肾血流。当内源性肾脏PG被激活,肾皮质内带和髓旁血流增加,但皮质外带血流改变不明显,这种现象,不仅见于PGE2,也见于其他血管舒张作用的PG。PGE2虽然可以增加肾血流,但不改变GFR,这种情况下入球动脉阻力可下降50%,出球动脉下降30%。应用PG抑制剂,可以抵销这些改变。血管收缩剂也可以引起代偿性的肾内合成PG增加。因此,各种因素减少肾脏血液动力学时,PG对维持肾脏血液动力学稳定有重要作用。
如上所述,血管紧张素Ⅱ(AⅡ)对肾小球滤过率的几个决定因素如血浆流量、入球动脉和出球动脉阻力以及肾小球滤过面积有重要作用。PGE2、PGI2对肾小球功能同样有重要作用,其作用是对肾脏内AⅡ相对抗。AⅡ在肾内受PG对抗,另一方面,AⅡ也刺激肾脏合成PG。对于AⅡ刺激作出反应释放出之PGE2,PGI2可以直接作用于肾小球感受器,特别是系膜细胞,增加细胞内cAMP,对系膜起调控作用。
如前所述,AⅡ主要作用部位在系膜,导致系膜细胞收缩,引起肾小球滤过面积缩小,PGE2有对抗AⅡ的这种作用。同样理由,环氧化酶抑制剂如消炎痛则强化AⅡ的这种作用。如果AⅡ作用被Saralasin抑制,则PGE2和PGI2舒张血管作用增加。
了解PG和血管紧张素肾内作用有重要意义。充血性心力衰竭、一些肝脏疾病、肾小球疾病、血容量不足等情况都伴有血浆肾素活性和AⅡ浓度增加,同时PG合成也增加。此时如果肾脏PGE2这种代偿性增加被阻断,如用消炎痛一类药物,则肾小球滤过率和肾血浆流量会急剧恶化。这种下降不仅受AⅡ作用,同时还受α-肾上腺素能儿茶酚胺以及加压素调节。但AⅡ受体此时如果也被阻断,则能减少该PG抑制剂的这种使肾功能恶化作用。动物实验,消炎痛引起的肾缺血,可以用Captopril处理而恢复。又如门脉高压、低钠。因PG抑制剂而GBF下降,如果用Captopril抑制AⅡ,肾血流可恢复。总之,无论如何,只要肾血管收缩因素,包括AⅡ,α-肾上腺素能儿茶酚胺及加压素增加,肾脏功能使成为“PG依赖”,抑制PG,则加强肾血管和肾小球毛细血管收缩,使RBF和GFR下降。此时血管紧张素转换酶抑制剂,在理论上有很好效益,临床上值得进一步观察
第四节 肾缺血机制的一些新概念
关于缺血性急诊肾功能衰竭(ARF)的发生机制,目前还不很清楚,根据近年来的研究,对肾内血管因素,肾内血流分布,以及肾小管功能损害等方面,有一些新的看法,在理论上,有重要意义。
一、血管因素
缺血性ARF时,可见总的肾血流量明显降低。在全身循环状况已经纠正后,这种降低常不立即消失,仍持续存在一段时间。因此,ARF时肾血流减少,可能有多种因素。由于血浆内肾素-血管紧张素浓度比此阶段明显增高,其活性增加,使人曾设想该因素引起缺血性ARF。但临床实验,并不支持此一简单看法。因为其他因素,如充血性心力衰竭、肝硬化、原发性高血压等,肾素活性也增加,但并不出现ARF。而且现已知肾内和血液中AⅡ活性增加,常常不在ARF之前出现,而在ARF之后。这些亦提示缺血性ARF时肾素活性增高,不是致病原因,而是肾缺血的结果。此外,如前所述,肾脏是产生PG的主要器官,与肾脏活动有关的,主要是PGE2、PGI和血栓素TXA2,前两者作用于血管相应受体,使血管舒张,阻力下降,而TXA2为缩血管物质,引起入球动脉收缩,但主要也是发生于肾小管阻塞以后。PGE2、PGI2的对抗AⅡ作用,有重要意义,动物实验,当给予动物外源性PGE2,肾缺血可减轻,肾皮质血流明显增加,如果PG合成受到抑制剂抑制,肾脏血管自动调节能力明显减弱。
二、肾血流分布
根据目前对肾缺血的研究,认为ARF时,并不支持以往看法,即肾皮质有严重缺血,而认为肾髓质在ARF时有明显淤血。在该情况下,病理损害主要表现在外髓层和皮质内层。由于肾脏微结构的不同,肾缺血时,外皮质肾单位的血流量和小管内流率改变不大,但髓旁肾单位的血流率有明显降低。如前所述,髓旁肾单位的出球动脉供应髓质血流,在此情况下,髓质供血降低,同时由于外髓层毛细血管的高度分支,使血液流变学易于改变,红细胞易于淤积,导致血管阻塞。形态学上已证实外髓内带和内髓区整个血管腔内血液淤滞。
此种现象机制不清楚,有人认为该部位亨利襻小管易于梗阻后扩张,压迫周围小静脉,以致淤血。另有人认为血管淤滞在前,影响对小管供血,引起小管损害。也可能两者互为因果。这种淤血程度和肾功能降低程度呈正相关。如果淤血持续存在,肾功能进行性恶化。
三、肾小管损害因素
肾脏在正常时,血流量很高,和其本身耗氧不成比例。如从肾静脉取血,可见其氧张力高于其他器官静脉血的含量。肾脏高血流量但又对血容量减低十分敏感,易于出现损害,其机制不十分清楚。近年来的研究展示氧输送到肾脏的分布复杂,各部位并非均一,呈梯度分布,因而局部的易损性可能由于选择性局部低氧所致。特别是外髓层近内髓层区,该部分小管即亨利襻厚升支段(mTAL)有主动吸收氯化钠和稀释尿液等作用,由于髓质部分的逆流交换系统和氯化钠的主动吸收,该处形成高代谢状态,使mTAL易受缺氧损害。当小管主动转运功能减低时,相对耗氧减少,则小管易受损程度减轻。如用Ouabain抑制细胞转运,或停止肾小球滤过,可以防止mTAL在游离灌注时肾缺血损害。反之,当膜转运增加,激化钠泵,可以使mTAL在同样条件下出现损害。因此,外髓质类似心绞痛综合征,即细胞缺氧程度依其氧需求和供氧两方的情况而定。按外髓层氧需求和供(血)氧的平衡观点,在低血压时,早期皮质流量减少,GFR下降,可以看作是降低髓质功能,使氧消耗减少,从而减轻或防止肾小管损伤,因此是一种保护作用。当此调节机制失效,mTAL缺血发生,其中溶质到达致密斑,启动管球反馈,GFR进一步下降。即进一步减少小管回吸收,减少耗氧。如果仍不足以保护mTAL缺血损害,则出现ARF。因此mTAL的损害,是ARF的中心环节,从选择性mTAL缺血到广泛灶性坏死,可能是临床上从轻型ARF到严重尿闭ARF的主要机制。