第二节 补体系统的激活
补体系统各成分通常多以非活性状态存在于血浆之中,当其被激活物质活化之后,才表现出各种生物学活性。补体系统的激活可以从C1开始;也可以越过C1、C2、C4,从C3开始。前一种激活途径称为经典途径(classical pathway)或替代途径。“经典”,“传统”只是意味着,人们早年从抗原体复合物激活补体的过程来研究补体激活的机制时,发现补体系统是从C1开始激活的连锁反应。从种系发生角度而言,旁路途径是更为古老的、原始的激活途径。从同一个体而言,在尚未形成获得性免疫,即未产生抗体之前,经旁路途径激活补体,即可直接作用于入侵的微生物等异物,作为非特异性免疫而发挥效应。由于对旁路途径的认识,远远晚在经典之后,加上人们先入为主观念,造成了命名的不合理。
一、经典激活途径
参与补体经典激活途径的成分包括C1~C9。按其在激活过程中的作用,人为地分成三组,即识别单位(Clq、Clr、Cls)、活化单位(C4、C2、C3)和膜攻击单位(C5~C9),分别在激活的不同阶段即识别阶段、活化阶段和膜功击阶段中发挥作用。
(一)识别阶段
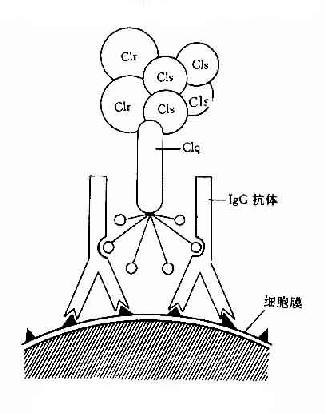
图3-1 CIq示意图
C1与抗原抗体复合物中免疫球蛋的补体结合点相结合至C1酯酶形成的阶段。
C1是由三个单位Clq、Clr和Cls依赖Ca+结合成的牢固的非活性大分子。
Clq:Clq分子有6个能与免疫球蛋白分子上的补体结合点相结合的部位。当两个以上的结合部位与免疫球蛋白分子结合时,即Clq桥联免疫球蛋白之后,才能激活后续的补体各成分(图3-1)IgG为单体,只有当其与抗原结合时,才能使两个以上的IgG分子相互靠拢,提供两个以上相邻的补体结合点不能与Clq接触,只有当IgM与抗原结合,发生构型改变,暴露出补体结合部位之后,才能与Clq结合。一个分子的IgM激活补体的能力大于IgG。Clq与补体结合点桥联后,其构型发生改变,导致Clr和Cls的相继活化。
Clr:Clr在C1大分子中起着连接Clq和Cls的作用。Clq启动后可引起Clr构型的改变,在活性的Clr,后者可使Cls活化。
Cls:Clr使Cls的肽链裂解,其中一个片段Cls具有酯酶活化,即CI的活性。此酶活性可被C1INH灭活。
在经典途径中,一旦形成Cls,即完成识别阶段,并进入活化阶段。
(二)活化阶段
CI作用于后续的补体成分,至形成C3转化酶(C42)和C5转化酶(C423)的阶段。
C4:C4是CI的底物。在Mg2+存在下,CI使C4裂解为C4a和C4b两个片段,并使被结合的C4b迅速失去结合能力。CI与C4反应之后能更好地显露出CI作用于C2的酶活性部位。
C2:C2虽然也是CI的底物,但CI先在C4作用之后明显增强了与C2的相互作用。C2在Mg2+存在下被CI裂解为两个片段C2a和C2b。当C4b与C2a结合成C4b2b(简写成C42)即为经典途径的C3转化酶。
C3:C3被C3转化酶裂解在C3a和C3b两个片段,分子内部的疏酯基(-S-CO-)外露,成为不稳定的结合部位。硫酯基经加水分解,成为-SH和-COOH也可与细菌或细胞表面的-NH2和-OH反应而共价结合。因此,C3b通过不稳定的结合部位,结合到抗原抗体复合物上或结合到C42激活C3所在部位附近的微生物、高分子物质及细胞膜上。这点,对于介导调理作用和免疫粘附作用具有重要意义。C3b的另一端是个稳定的结合部位。C3b通过此部位与具有C3b受体的细胞相结合(图3-2)。C3b可被I因子灭活。C3a留在液相中,具有过敏毒素活性,可被羟肽酶B灭活。
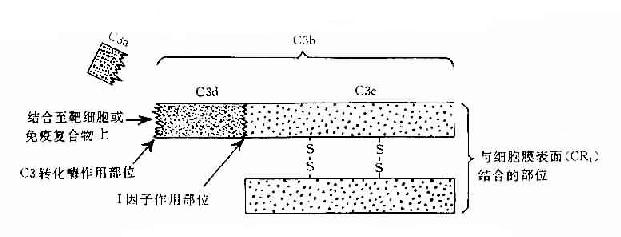
图3-2 C3分子及其裂解产物生物活性示意图
C3b与C42相结合产生的C423(C4b2b3b)为经典途径的C5转化酶。至此完成活化阶段。
(三)膜攻击阶段
C5转化酶裂解C5后,继而作用于后续的其他补体成分,最终导致细胞受损、细胞裂解的阶段。
C5:C5转化酶裂解C5产生出C5a和C5b两个片段。C5a游离于液相中,具有过敏毒素活性和趋化活性。C5b可吸附于邻近的细胞表面,但其活性极不稳定,易于衰变成C5bi。
C6~C9:C5b虽不稳定,当其与C6结合成C56复合物则较为稳定,但此C5b6并无活性。C5b6与C7结合成三分子的复合物C5b67时,较稳定,不易从细胞膜上解离。
C5b67即可吸附于已致敏的细胞膜上,也可吸附在邻近的,未经致敏的细胞膜上(即未结合有抗体的细胞膜上)。C5b67是使细胞膜受损伤的一个关键组分。它与细胞膜结合后,即插入膜的磷脂双层结构中。
若C5b67未与适当的细胞膜结合,则其中的C5b仍可衰变,失去与细胞膜结合和裂解细胞的活性。
C5b67虽无酶活性,但其分子排列方式有利于吸附C8形成C5678。其中C8是C9的结合部位,因此继续形成C5~9,即补体的膜攻击单位,可使细胞膜穿孔受损。
目前已经证明,不C5b、C6、C7结合到细胞膜下是细胞膜仍完整无损;只有在吸附C8之后才出现轻微的损伤,细胞内容物开始渗漏。在结合C9以后才加速细胞膜的损伤过程,因而认为C9是C8的促进因子。(图3-3)。

图3-3 经典途径的激活
二、旁路激活途径
旁路激活途径与经典激活途径不同之处在于激活是越过了C1、C4、C2三种成分,直接激活C3继而完成C5至C9各成分的连锁反应,还在于激活物质并非抗原抗体复合物而是细菌的细胞壁成分—脂多糖,以及多糖、肽聚糖、磷壁酸和凝聚的IgA和IgG4等物质。旁路激活途径在细菌性感染早期,尚未产生特异性抗体时,即可发挥重要的抗感染作用。
(一)生理情况下的准备阶段
在正常生理情况下,C3与B因子、D因子等相互作用,可产生极少量的C3B和C3bBb(旁路途径的C3转化酶),但迅速受H因子和I因子的作用,不再能激活C3和后续的补体成分(图3-4,左)。只有当H因子和I因子的作用被阻挡之际,旁路途径方得以激活(图3-4,右)。
C3:血浆中的C3可自然地、缓慢地裂解,持续产生少量的C3b,释入液相中的C3b迅速被I因子灭活。
B因子:液相中缓慢产生的C3b在Mg2+存在下,可与B因子结合形成C3Bb。
D因子:体液中同时存在着无活性的D因子和有活性的D因子(B因子转化酶)。D因子作用于C3bB,可使此复合物中的B因子裂解,形成C3bBb和Ba游离于液相中。C3bBb可使C3裂解为C3a和C3b,但烊际上此酶效率不高亦不稳定,H因子可置换C3bBb复合物中的Bb,使C3b与Bb解离,解离或游离的C3b立即被I因子灭活。因此,在无激活物质存在的生理情况下,C3bBb保持在极低的水平,不能大量裂解C3,也不能激活后续补体成分。但是这种C3的低速度裂解和低浓度C3bBb的形成,具有重大意义。可比喻为处于“箭在弦上,一触即发”的状态。
(二)旁路途径的激活
旁路途径的激活在于激活物质(例如细菌脂多糖、肽聚糖;病素感染细胞、肿瘤细胞,痢疾阿米巴原虫等)的出现。目前认为,激活物质的存在为C3b或C3bBb提供不易受H因子置换Bb,不受Ⅰ因子灭活C3b的一种保护性微环境,使旁路激活途径从和缓进行的准备阶段过渡到正式激活的阶段(图3-4)。

·

图3-4 旁路途径的激活
左:在正常后理情况下,可产生出少量C3bBb,但迅即被激活。
右:在激活物存在下,C3b不易被I因子灭活,C3bBb中的Bb不易被H因子置换,使激活过程得以进行。
P因子:P因子旧称备解素(properdin)。正常血浆中也有可以互相转换的两种P因子,P和P。C3bBb的半衰期甚短,当其与P因子结合成为C3bBbP时,半衰期可延长。这样可以获得更为稳定的、活性更强的C3转化酶。
C3bBb3b:C3bBb与其裂解C3所产生的C3b可进一步形成多分子复合物C3bBb3b。C3bBb3b像经典途径中的C5转化酶C423一样,也可使C5裂解成C5a和C5b。后续的C6~C9各成分与其相互作用的情况与经典途经相同。
(三)激活效应的扩大
C3在两条激活途径中都占据着重要的地位。C4是血清中含量最多的补体成分,这也正是适应其作用之所需。不论在经典途径还是在旁路途径,当C3被激活物质激活时,其裂解产物C3b又可在B因子和D因子的参与作用下合成新的C3bBb。后者又进一步使C3裂解。由于血浆中有丰富的C3,又有足够的B因子和Mg2+,因此这一过程一旦被触发。就可能激活的产生显著的扩大效应。有人称此为依赖C3Bb的正反馈途径,或称C3b的正反馈途径(图3-5)。
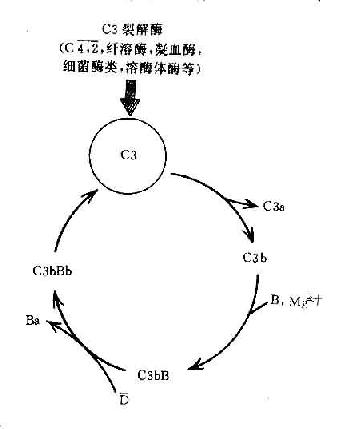
图3-5 C3b的正反馈途径
三、两条激活途径的比较
补体的两条激活途径有共同之处,又有各自的特点。在补体激活过程中,两条途径都是补体各成分的连锁反应,许多成分在相继活化后被裂解成一大一小两个片段;不同的片段或片段的复合物可在靶细胞表面向前移动,如C42,C423,C5b,C567,虽亦可原始的激活部位就地形成复合物,但仍以移动为主,在激活过程中,补体成分和(或)其裂解产物组成更大的复合物,同时又都在扩大其激活效应,这一过程可形象地比喻为“滚雪球”。
两条途径的不同之处参见表3-4及图3-6。
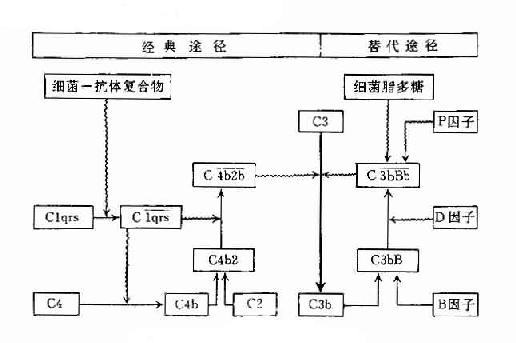
图3-6 两条激活途径的比较
表3-4 两条激活途径的主要不同点
| 比较项目 | 经典活途径 | 旁路激活途径 |
| 激活物质 | 抗原与抗体(IgM、IgG3、IgG1、IgG2)形成的复合物 | 细胞脂多糖、凝聚的IgG、IgA等 |
| 参与的补体成分 | C1~C9 | C3,C5~C9,B因子,D因子,P因子等 |
| 所需离子 | Ca2+,Mg2+ | Mg2+ |
| C3转化酶 | C42(C4b2b) | C3bBb |
| C5转化酶 | C423(C4b2b3b) | C3bBb3b |
| 作用 | 参与特异性体液体免疫的效应阶段 | 参与非特异性免疫,在感染早期即发挥作用 |
四、补体激活过程的调节
机体通过一系列的复杂的因素,调节补体系统的激活过程,使之反应适度。例如经C3b的正反馈途径即可扩大补体的生物学效应。但补体系统若过度激活,不仅无益地消耗大量补体成分,使机体抗感染能力下降;而且在激活过程中产生的大量行物活性物质,会使机体发生剧烈的炎症反应或造成组织损伤,引起病理过程。这种过度激活及其所造成的不良后果,可通过调控机制而避免。这种调控机制包括补体系统中某些成分的裂解产物易于自行衰变以及多种灭活因子和抑制物的调节作用。
(一)自行衰变调节
某些补体成分的裂解产物极不稳定,易于自行衰变,成为补体激活过程中的一种自控机制。例如C42复合物中的C2b自行衰变即可使C42不再能持续激活C3,从而限制了后续补体成分的连锁反应。C5b亦易于自行衰变,影响到C6~C9与细胞膜的结合。
(二)体液中灭物质的调节
血清中含有多种补体成分的抑制或灭活特定的补体成分。
CI抑制物:CI抑制物(Ci inhibitor,CIINH)可与CI不可逆地结合,使后者失去酯酶活性,不再裂解C4和C2,即不再形成C42(C3转化酶),从而阻断或削弱后续补体成分的反应。遗传性CIINH缺陷的患者,可发生多以面部为中心的皮下血管性水肿,并常以消化道或呼吸道粘膜的局限性血管性水肿为特征。其发生机制是CI未被抑制,与C4、C2作用后产生的C2a(旧称C2b的小片段)为补体激肽,或增强血管通透性,因而发生血管性肿。
CIINH缺陷时,C4、C2接连不断地被活化,故体内C4、C2水平下降;因其不能在固相上形成有效的C42(C3转化酶),所以C3及其后续成分不被活化。因此本病不像C3~C8缺陷那样容易发生感染。
大部分CIINH缺陷病人与遗传有关,另有约15%的病人无遗传史,其CIINH虽有抗原性但无活性(部分可产生正常CIINH,并非完全缺陷)。前者称为I型血管性水肿,后者称为Ⅱ型血管性水肿(Alsenz等,1987)。
血管性水肿可用提纯的CIINH治疗,据称有效,亦可给以男性激素制剂以促进肝合成CIINH,预防水肿的发生。
C4结合蛋白:C4结合蛋白(C4 binding protein, C4bp)能竞争性地抑制C4b与C2b结合,因此能抑制C42(C3转化酶)的形成。
I因子:I因子又称C3b灭活因子(C3b inactivator, C3b INA)能裂解C3b,使其成为无活性的C3bi,因而使C42及C3bBb失去与C3b结合形成C5转化酶的机会。
当遗传性I因子缺陷时,C3b不被灭活而在血中持续存在,可对旁路途径呈正反馈作用,陆续使C3裂解并产生出更多的C3b。因此血中C3及B因子的含量因消耗而降低。当发生细菌性感染时,因补体系统主要成分C3和B因子严重缺乏,削弱了抗感染作用,可因条件致病菌惹发严重的甚至致命性后果。
H因子:H因子虽能灭活C3b,但不能使C3bBb中的C3b灭活。H因子(factor H)不仅能促进I因子灭活C3b的速度,更能竞争性地抑制B因子与C3b的结合,还能使C3b从C3bBb中致换出来,从而加速C3bBb的灭活。由此可见,I因子和H因子在旁路途径中,确实起到重要的调节作用。
S蛋白:S蛋白(Sprotein)能干扰C5b67与细胞膜的结合。C5b67虽能与C8、C9结合,但它若不结合到细胞膜(包括靶细胞的邻近的其他细胞)上,就不会使细胞裂解。
C8结合蛋白:C8结合蛋白(C8binding protein,C8bp)又称为同源性限制因子(homologousrestriction factor,HRF)。C56与C7结合形成C567即可插入细胞膜的磷脂双层结构之中,但两者结合之前,可在体液中自由流动。因此,C567结合的细胞膜不限于引起补体激活的异物细胞表面,也有机会结合在自身的细胞上,再与后续成分形成C5~9大分子复合物,会使细胞膜穿孔受损。这样会使补体激活部位邻近的自身细胞也被殃及。
C8bp可阻止C5678中的C8与C9的结合,从而避危及自身细胞膜的损伤作用。C8分子与C8bp之间的结合有种属特异性,即C5678中的C8与同种C8bp反应;但与异性种动物的C8不反应,所以又称为HRF。据称C8bp也能抑制NK细胞和Tc细胞的杀伤作用,值得注意。