��һ�ڡ�����ͻ���µ����ʺϳ��쳣
��������������DNA�����ϼ��������˳������ġ����DNA���ӵļ��������˳�����仯����������ĵ����ʽṹ�ͷ�����Ӧ�ĸı䡣���ڻ���ͻ�䵼�µ����ʷ����ʺ����쳣���Ӷ�������幦���ϰ���һ�༲����Ϊ���Ӳ���molecular disease����
���Ӳ�����ܶ�,���ݸ��ֵ����ʵĹ��ܿɽ����Ӳ���Ϊ�����Ե��ײ�����Ѫ������Ѫ����ȱ��֢�����ߵ���ȱ�ݲ���Ĥ���ײ������嵰�ײ���.
һ��Ѫ�쵰�ײ�
Ѫ�쵰�ײ���hemoglobinopathy����ָ�����鵰���ӽṹ��ϳ����쳣������ļ��������������ϵ¶����Ŵ������о��������롢�����ķ��Ӳ����������Ե��ײ��Ĵ��������о������Ŵ����������ģ�͡��ݹ��ƣ�ȫ������һ�ڶ���Я��Ѫ�쵰�ײ��Ļ����ҹ��Ϸ������ʽϸߣ���ˣ�Ѫ�쵰�ײ���������Ŵ�֮һ��
��һ������Ѫ�쵰����ɣ��ṹ���Ŵ�����
1������Ѫ�쵰����ɺͷ����仯 ÿ����ϸ���ں���Լ28000���Ѫ�쵰���ӣ�ÿ���������ĸ��ǵ�λ���ɣ�ÿһ����λ��һ���鵰��������һ��Ѫ���ظ�����ɣ���Ѫ�쵰�������ɶ����鵰�������ɵ������ľ��壨ͼ4��10��������һ����������������ͦ���������141����������ɣ���һ���������(�š��¡��úͦ���)����146����������ɡ�����6�ֲ�ͬ���鵰������ϳ������6�ֲ�ͬ��Ѫ�쵰�ף���Hb Gower1(��2��2)��HbGower2��(��2��2)��HbPortland(��2��2)��HbF(��2��2)��HbA(��2��2)��HbA2(��2��2)�����Ц������������ͣ���G��2��A��2�����HbF�����ࣺ��2G��2�ͦ�2A��2��ǰ�ߵĵ�136λ����Ϊ�ʰ��ᣬ����Ϊ�����ᡣ
��������Ѫ�����ڷ����IJ�ͬ���Ⱥ�����֣�ͼ4��11��������̥�������ڣ��ϳ���̥Ѫ�쵰��HbGowerl��HbGower2��HbPortland��̥���ڣ���8��������Ϊֹ����Ҫ��HbF��������3��Ѫ�쵰�ף�HbA��ռ95�����ϣ�HbA2��ռ2%-3.5%��HbF������1.5%��
2�������鵰���������鵰�����Ϊ���ࣺһ��������鵰����أ���-likeglobin gene cluster���������κͦ�������һ���Ǧ��鵰����أ���-like globin gene cluster��,�����š��ã�G�ú�A�ã����ĺͦ»���
��1������鵰����������鵰�����λ��16p13��ÿ��Ⱦɫ���Ͼ����������鵰������ˣ�������ϸ���й���4��������ÿ�����������������Ħ��鵰���������⣬������鵰������У������������λ����һ���ٻ�������Щ�����������������˳����ͼ4��12��ʾ��
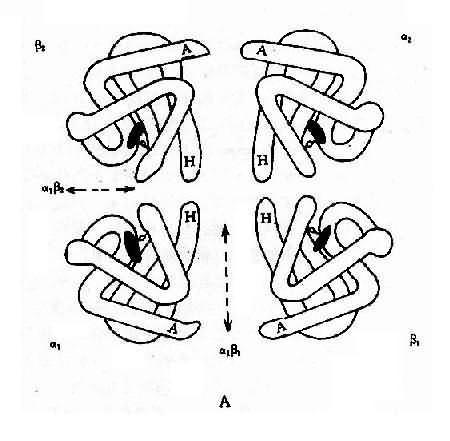
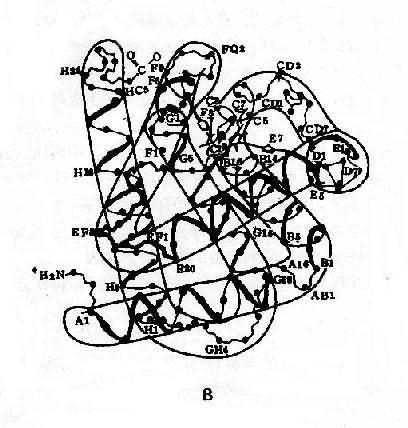
ͼ4��10 Ѫ�쵰�ṹʾ��ͼ
A��Ѫ�쵰���ľ��壨�ļ��ṹ��
B��Ѫ�쵰�������ά�ռ�ṹ
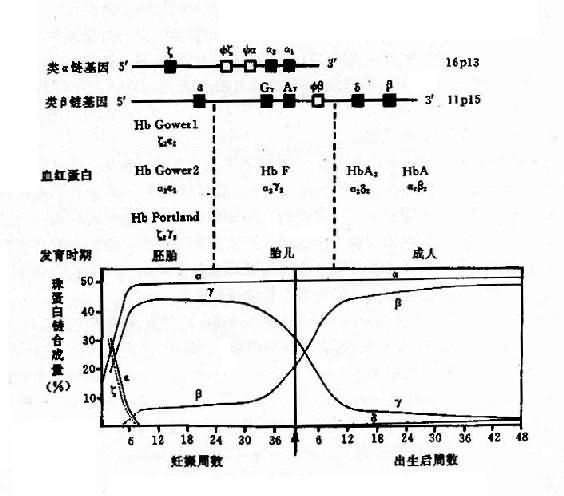
ͼ4��11 ����Ѫ�쵰�����ͼ������������е��ݱ�
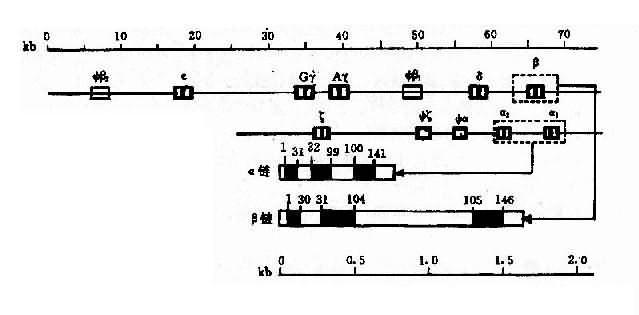
ͼ4��12 ������鵰����غ�������鵰����صĽṹ������˳��
��2������鵰����������鵰����طֲ���11p15��ÿ��11��Ⱦɫ����ֻ��һ�����鵰��������ڸ���ԱҲ������������������˳����ͼ4��12������鵰����ص�����˳�������������еı��������ȫһ�¡�
��3���鵰����Ľṹ�����������鵰����Ľṹ���ƣ�������3�������Ӻ�2���ں��ӣ�IVS2��IVS2�������鵰�����е�IVS1��117bp��ɣ�λ��31��32������֮�䣬IVS2��149��140bp��ɣ�λ��99��100������֮�䡣��»����е�IVS1��130bp��λ��30��31����֮�䣬IVS2��Լ��850bp��λ��104��105������֮�䡣
������Ѫ�쵰�ײ��ķ���ͷ��ӻ���
Ѫ�쵰�ײ��ɷ�Ϊ�����࣬���쳣Ѫ�쵰�ײ��͵��к�ƶѪ��
1���쳣Ѫ�쵰�ײ� �쳣Ѫ�쵰�ף�abnormalhemoglobin����ָ�����鵰����ͻ�䵼���鵰�������ṹ�쳣�������ٴ������߳�Ϊ�쳣Ѫ�쵰�ײ����쳣Ѫ�쵰���ۺ���������ȫ�����ѷ����쳣Ѫ�쵰��471�֡������ѷ���60�֣�����20���������ױ��������쳣Ѫ�쵰������࣬����Լ40�����쳣Ѫ�쵰�������в�ͬ�̶ȵĹ����ϰ���
��1���쳣Ѫ�쵰�ײ������ͣ�
1������ϸ������sickle cell disease�����˲���Ҫ���ں��ˡ��ò�ϵ���ڦ�����6λ�Ȱ��ᱻ�Ӱ���ȡ�����γ�HbS�����µ�ɸı䣬�����������HbS�ۺ��γɳ���״�ۺ��ʹ��ϸ�����䣬������������Ѫճ�����ߣ�����Ѫ�ܹ����Լ̷�֢״��һ���Ծ�ʹ���������ʹ����ʹ�������Դ������֯���ˣ��ļ�����������������ϸ���ı��Խ��ͻ���������Ѫ��HbS�����ӣ�HbSHbS������Ϊ����ϸ����ƶѪ���Ӻ��ӣ�HbAHbS������Ϊ����ϸ����״������֢״����Ҳ�����������ƶѪ��
2�����ȶ�Ѫ�쵰�ײ���unstable hemoglobinpathies��:�ѷ��ֵIJ��ȶ�Ѫ�쵰����80�����ϡ�����Hb���ȶ������Է������������������£����ԣ��γɱ����鵰��С�壨HeinzС�壩��HeinzС��ճ����ϸ��Ĥ�ϣ�����������ͨ�����ӣ����⣬���ڱ����Խ��ͣ�����ϸ��ͨ��ѭ��ʱ����ϸ���������ƻ�������Ѫ���ڡ�����Ѫ�����ȶ�Hb��һ��ʳ�Ⱦɫ�������Ŵ�������ȫ���ԣ����Ӻ��ӿ����ٴ�֢״�������ӿ��������ٴ�������Hb���ȶ��̶ȡ���������Ѫ�쵰�Ķ����Լ����ȶ�Hb����������С�йء����߽��ڷ��ûǰ���ҩ����и�Ⱦʱ��Ѫ�������跴����Ѫ����ά��������
3��Ѫ�쵰�ףͲ���HbM����HbM������������Ѫ������ԭ�����ӵ��鰱����ڽ��İ����ᷢ������������²�����ԭ�ӳ��ȶ��ĸ���״̬���Ӷ�Ӱ���������Ĵ������ܣ�ʹ��֯�������㣬�����ٴ��ϳ�����示ͼ̷��Ժ�ϸ�����ࡣ�����ʳ�Ⱦɫ�������Ŵ����Ӻ���HbM����һ����30�����ڣ����������֢״��
4���������ı��Ѫ�쵰�ײ�������Ѫ�쵰�ײ���ָ���������ϰ����������ʹѪ�쵰�����������������ͣ������������ܸı䡣������Hb�����������ߣ�������֯���������٣����º�ϸ������֢��������Hb�����������ͣ���ʹ����Ѫ�������Ͷ��½��������߿��������֢״��
��2���쳣Ѫ�쵰�ķ��ӻ������쳣Ѫ�쵰�ķ����漰����ͻ��ĸ������ͣ������������¡�
1����������û���������쳣Ѫ�쵰���������鵰��������������û����£����ж�Ϊ����ͻ�䡣
�ٴ���ͻ�䣺��������ϸ��ƶѪ�Ǧ»����6λ������GAG���GTG���й��˽ϳ�����HbE�Ǧ»����26λ��������GAG���ȣ���AAG���������¡�
������ͻ�䣺����HbMckees-Rock,�����ֻ��144����������ɣ�ԭ���Ǧ»����145λ�Ұ���������TAT�ı�Ϊ��ֹ������TAA��ʹ�����ϳ���ǰ��ֹ��
����ֹ����ͻ�䣺����Hb Constant Spring�������ڦ��鵰�����142λ��ֹ������TAA��mtRNAΪUAA��ͻ��ΪCAA���Ȱ���������������ӳ�Ϊ172�������ᣬ����ͻ�����ת�γɵ�mRNAI���ȶ������Ե��¦����ϳɼ��٣�����Ϊ�������к�ƶѪ��
2������ͻ�䣺����Hb Wagne�����ڦ�����138λ˿����������UCC��ʧһ��C����ʹ��3���˼��˳������λ�ƣ����±��룬��142λ��ֹ�źű�Ϊ�ɶ����룬��ʹ������147λ����ֹ��ͼ4��13����
3������ͻ�䣺����Hb Gum Hiu�Ǧ���ȱʧ��91��95�����ᣨ�����飭���ף��Ŷ�������������ǰ����˳��������ͼ4��13����

ͼ4��13 ��������ͦ���mRNAR��ͬͻ������
4���ںϻ�������Hb Lepore����������ɦ����ͦ������Ӷ��ɣ�������N���������C����������ʳ�Ϊ�Ħ������෴��Hb��Lepore(Hbanti-Lepore)��N���������C��ȴ���������Ϊ�¦�������������Ⱦɫ��Ĵ�������Ͳ��Ƚ������γɵ��ںϻ���fusion gene��(ͼ4��14)����
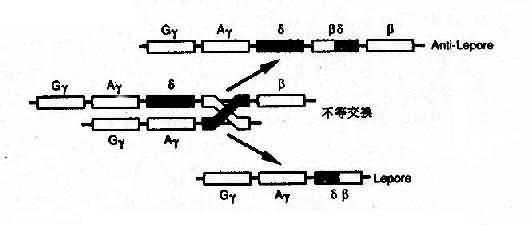
ͼ4��14 Ѫ�쵰���ںϻ����γɻ���
2�����к�ƶѪ�����鵰����ȱʧ��ͻ�䵼��ij���鵰�����ϳ��ϰ�����ɦ����ͦ����ϳ�ʧȥƽ���浼�µ���Ѫ��ƶѪ��Ϊ���к�ƶѪ��thalassemia�������ݺϳ��ϰ���������ͬ�ɰѵ��к�ƶѪ��Ϊ���ͦµ��к�ƶѪ���ࡣ������ټ��ĦĦºͦæµ��к�ƶѪ��
��1�������к�ƶѪ����-thalassemia,��Ʀ���ƶ�������ڦ��鵰�����ȱʧ��ȱ��ʹ���鵰��������Ʀ������ĺϳ��ܵ����ƶ��������Ѫ��ƶѪ�����һ��16��Ⱦɫ��ȱʧ1���������߳Ʀ�����ƶ����Ʀ���2����ȱʧ2���������߳�Ϊ��0��ƶ����Ʀ���1��������1�Ļ����Ϳ�д���D������������2�ĵ�������д�ɦ�-/����.����ÿ�֦���ƶ����������������Ͽɹ��ɸ��֦���ƶ�Ӻ���.���֦���ƶ�������Ӻ������Ͽɹ��ɸ��ִ����ӻ�˫���Ӻ���.�����к�ƶѪ���ҹ�������Ϸ���ʡ.
1)����ƶ���ٴ�����:�����ٴ����̶ֳ�,�����ۦ�����������ͬ���в���,�����Ͽɷ�Ϊ4����.
��HbBart��s̥��ˮ���ۺ���(HbBart��s hydrops fetalis syndrome):������16Ⱦɫ���4��������ȫ��ȱʧ��ȱ��,������Ϊ��0��ƶ������(--/--),��ȫ���ܺϳɦ���,�����γ�̥��HbF,��Թ���Ħ����γɦ��ľ���(��4)��ΪHbBart��s(��4).HbBart��s���������dz���,����ͷŸ���֯��������,�����֯����ȱ������̥��ˮ��,������̥������������.����Ѫ�쵰��60%����ΪHbBart��s,����Ϊ HbPortland.
������ĸ��Ϊ��0��ƶ�Ӻ���,������Ϊ��/--.������������,��̥����1/4�Ļ���Ϊ��HbBart��sˮ��̥��,1/4Ϊ������,1/2Ϊ��0��ƶ�Ӻ���(����1)
��Ѫ�쵰��H�����Ǧ�0��ƶ�ͦ�+��ƶ��˫���Ӻ��ӣ�����3��������ȱʧ��ȱ�ݣ�������Ϊ-��/--���-/--,Ҳ��Ϊ����T/--(��T������ͻ��,��Hb Constant Spring)����ȱʧ3��������ֻ�ܺϳ�����������������Թ��࣬�γɦ��ľ���(��4)���ױ����������¦�4���������ĵ�����������������ۻ������壬�����ں�ϸ��Ĥ�ϣ�ʹ��ϸ��Ĥ����ʧȥ�����ԣ��ױ�Ƣ�ƻ��������еȶȻ�����ص���Ѫ��ƶѪ����ΪѪ�쵰��H����Hbh disease�� .
����˫�Ļ����Ͷ�Ϊ��0��ƶ�Ӻ��ӣ�����/--���ͦ�+��ƶ�Ӻ��ӣ���/����������Ϊ��0��ƶ�Ӻ��ӣ�����/--���ͷ�ȱʧ�͵�ƶ�Ӻ��ӣ�����T/�����������������Ů��1/4����Ϊ������1/4Ϊ��+��ƶ�Ӻ��ӣ�1/4Ϊ��0��ƶ�Ӻ��Ӽ�1/4ΪHbH�����縸ĸһ���Ц�T�����·�ȱʧ��HbH����
�����ͣ����� �������к�ƶѪ��Ϊ��0��ƶ�ӷ��ӣ�--/���������+��ƶ�Ӻ��ӣ���/��-����ȱʧ����������������ƶѪ���ҹ���Ҫ�Ǧ�0��ƶ�Ӻ��ӡ�
���ͦ���ƶ����֮����䣬������Ů�п���1/4����ΪHbBartˮ��̥���ۺ�����
�ܾ�ֹ�ͦ����к�ƶѪ����ȱʧһ��������Ϊ��+��ƶ�Ӻ��ӣ�-��/����������֢״��
��ֹ�ͦ���ƶ�����͵��к�ƶѪ������䣬����1/4��������HbH��������
2�������к�ƶѪ�ķ��ӻ������ӻ���ȱ�ݳ̶������֣��ɰѦ���ƶ��Ϊȱʧ�ͺͷ�ȱʧ�ͣ���ͻ�䣩��
�ٻ���ȱʧ���ɷ�Ϊ��+�ͦ�0��ƶ���֡���+��ƶ�����ֻ����ͣ����ȱʧ��leftward deletion��,ȱʧһ��������2�������ڵ�DNAƬ�Σ����Ҳ�ȱʧ��right ward deletion��, ȱʧ��Χ������2����3���˺ͦ�1�����5���ˣ�����γ����ɦ�1��3���˺ͦ�2��5���˹��ɵ��ںϻ����䷢��������������������Ƚ����Ľ������0��ƶ����ȱʧ��Χ���ܴ�ͼ4-15����

ͼ4-15 ��������ȱʧ����
�ڷ�ȱʧ�ͣ���ͻ�䣺�����ͼ���4-1����
��2���µ��к�ƶѪ���µ��к�ƶѪ���thalassemia,��Ʀµ�ƶ���������ڦ��鵰�����ȱʧ��ȱ��ʹ���鵰��������Ʀ������ĺϳ��ܵ����ƶ��������Ѫ��ƶѪ����ȫ���ܺϳɦ����߳Ʀ�0��ƶ���ܲ��ֺϳɦ����ߣ�ԼΪ������5%-30%���Ʀ�+��ƶ�����⣬���ЦĦµ�ƶ�����ǿ����в�ͬ����ϣ�����0��ƶ�����ӣ���0��0������0��ƶ˫���Ӻ��ӣ���0/��+������0��ƶ�Ӻ��ӣ���0��A������+��ƶ�����ӣ���+/��+���ͦ�+��ƶ�Ӻ��ӣ���+/��A�����µ�ƶ���ҹ��Ϸ��ϳ�����
1���ٴ����ࣺ���¿���4����Ҫ���͡�
�����ͦµ��к�ƶѪ�������Ǧ�+��ƶ����0��ƶ��Ħ�0��ƶ�Ĵ����ӣ�������ͷֱ�Ϊ��+/��+����0/��0�ͦĦ�0/�Ħ�0�����Ǧ�+�ͦ�0��ƶ��˫���Ӻ��ӣ�������Ϊ��0/��+������Щ���ߵĦ����������ܺϳɣ���ϳ������٣�������HbA�����ܵͣ������ĺϳ�������ӣ�ʹHbFt GbA2�������ߡ�����HbF��HbA���������ߣ�����֯�в����ͷų��������Цµ�ƶ��������֯ȱ��֢״����֯ȱ����ʹ��ϸ�������ش������ڣ��̼��������Ѫ���ܣ�ʹ���������������������ʴ�¹������ɣ��ɳ��֡����к�ƶѪ���ݡ���ͷ�������¡�𣬱������ݣ�������ǰͻ���۾�����������ף������ڦ����ϳ������ƣ���ʣ����������γɦ��������壬������Ѫ��ƶѪ������Ѫά��������
��4-1 ��ͻ������ĵ��к�ƶѪ
| ����ȱ������ |
| 1���������ܻ��ȶ��Խ��͵� ������ͻ�� a116GAG---TAG,(G---T) ������ͻ�� a130/31(--4bp) ����ֹ����ͻ�� 142TAA---CAA(T---C) �γ�Hb Constant Spring ����ʼ����ͻ�� a2ATG---ACG(T--C) 2��RNA�ӹ�ͻ�� �ټ��Ӹı� IVSI��GGTGAGGCT---GGCT�� ��Poly(A)�ź�ȱ�� AATAAAA ---AATAAG 3���������ȶ�H a2125CTG������CCG������ ����Hb Quong��Sze |
�����ͦµ��к�ƶѪ�������Ǧ�+��ƶ����0��ƶ��Ħ�0��ƶ���Ӻ��ӣ������ͷֱ�Ϊ��+/��A����0/��+�ͦĦ�0/��A����������ڻ��ܺϳ��൱���Ħ���������֢״���ᣬƶѪ�����Ի����ƶѪ�������ص���HbA2���ߣ��ɴ�4%-8%���ͣ�HbF���ߡ�
���м��ͦµ��к�ƶѪ������ͨ����ijЩ�µ�ƶ�����͵Ĵ����ӣ����+��ƶ����F��/��+��ƶ����F�������ֲ�ͬ�����͵�ƶ��˫���Ӻ��ӣ����+����ƶ/�Ħ�+��ƶ����֢״�������ͺ�����֮�䣬�ʳ�Ϊ�м��ͦµ��к�ƶѪ��
���Ŵ�̥��Ѫ�쵰�׳�������֢�����������ڦ»������ijЩDNAƬ�ε�ȱʧ���ߵ�ͻ�䣬ʹ�ĺͦ����ϳ������ƣ��������ĺϳ��������ӣ�ʹ���˺�ϸ����HbF�����������࣬�ʳ�Ϊ�Ŵ���̥��Ѫ�쵰�׳�������֢��hereditarypersistance of fetal hemoglobin,HPFH��.HPFH���ص���HbF�ij������Գ����ϸ�ˮƽ�������Ե��ٴ�֢״��
2���µ��к�ƶѪ�ķ��ӻ��µ��к�ƶѪ�����ѷ���100����ͻ�����ͣ�����10����Ϊȱʧ�ͣ������Ϊ��ͻ�䡣�ҹ��ѱ���17�ֵ�ͻ�䡣
��ͻ�䣺��������µ��к�ƶѪ�����ڦ»�������ͻ�����£�ͻ���漰�����ڼ��Բ����˳��ĸ������ڡ���Ҫ������4�֡�
a.������������ͻ�䡢����ͻ�����ʼ����ͻ�䣺ʹ���ɵ�mRNA�ȶ��Խ��ͻ��γ����ܵ�mRNA���Ӷ����ܺϳ������Ħ��鵰����������������0��ƶ������Ϊ��+��ƶ����������ͻ��������17��A��G����43��G��T����������0��ƶ������ͻ��41/42��-TCTT����71/71��+A���ͦ�0��ƶ���Լ���ʼ������ͻ��ATG��AGG���µĦ�0�������ࡣ�����й��ˡ�
b.�DZ�����IVS-1��IVS-2ͻ�䣺Ӱ��ǰmRNA���ӵȼӹ����̲���ȷ���У��γ��쳣��mRNA�����¦�0���+��ƶ������IVS-1��1λG��TΪRNAƴ�Ӵ��ı䣻�й����г�����IVS-2654��Ϊ�ں��������ڼ���û��γ���һ���µ��ѽ��źţ�Ӱ������λ��ļ��ӣ������쳣mRNA������һ�����ں����м���λ���ͨ��˳���ϵ�ͬ��ͻ�䣬�Ӷ������ں��ӻ��������������ѽ�λ�㣨cryptic splicihng site,CSS����IVS-15��G��C����CSS��DNA��һ��˳���ڵ�ͻ�������γɼ���ʶ��˳��CCTATTGGT���ĵ�7�����G���ΪA��������µ��е㣬��CCTATTAG��T����
c.Ӱ��ת¼��ͻ�䣺����ͻ����Ҫ��������ʼλ�����ε�������TATA��ʹת¼Ч�ʽ��ͣ�mRNA���������ٶ�������+��ƶ���й��˵�-29A��G��-28A��G���������ࡣ
d��RNA�ѽⲿλȱ�ݣ�����ͻ���������쳣��RNA��ñ��λ�Ͷ�������ữ�źŵ�ͻ�䣬�Ӷ�Ӱ��RNAת¼������ȷ�ѽ⣬�������ȶ���mRNA��ʹ�����������������٣����¦�+��ƶ��������mRNA��ñ��λ����A��C���������+��ƶ�������ˡ������磬��������ữ�ź�AATAAA��AACAAA�������+��ƶ��
e.��������������ͻ������������õĸı䣺����ͻ�������ڱ������ĵ����ͻ�䣨����ͻ���ͬ��ͻ�䣩�����ڽ��������ѽ��źţ�Ӱ��IVS����λ��ļ��ӣ������쳣��mRNA���綫���dz���HbE,��һ�����͵Ħµ�ƶ����ԭ���ǵ�����26λ�����ӷ���G��A����ͻ��ʱ�������ڵ��ѽ��źű���������쳣mRNA������HbE��ͼ4-16����
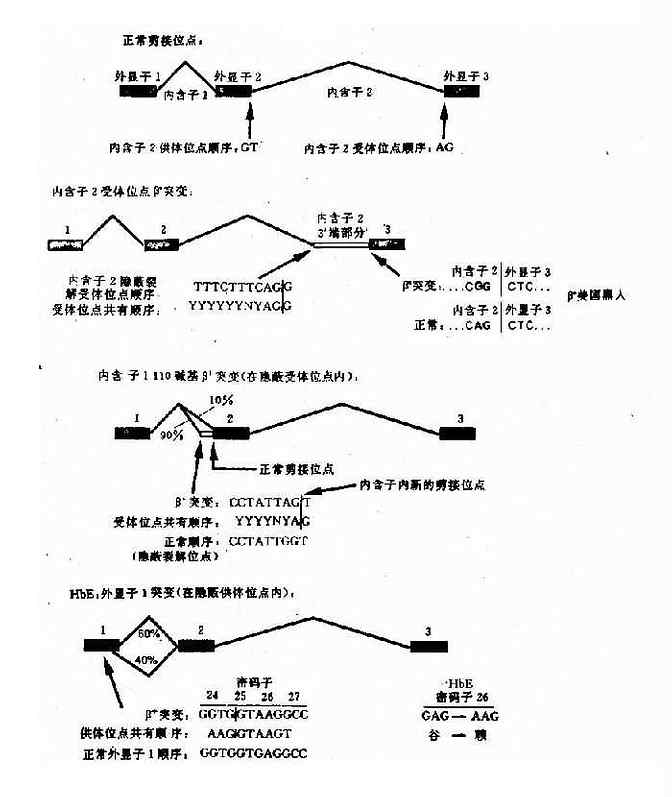
ͼ4-16 �����������鵰���ӵ�ͻ�����
HbE:������26��G-��A��
�ȡ���
GAG��AAG��QAJ��������ͻ�䣩
��+��HbA 60%�� ��E(HbE 40%)
3)��»���ȱʧ���ٰ�����鵰�����ȱʧ���̴��¿ɷ�Ϊ4�֣�����0���Ħ¡��æĵ��к�ƶѪ���Ŵ���̥��Ѫ�쵰�׳�������֢���ڵ������ڦ�0����ȱʧ����Ħµ��к�ƶѪ���������ںϻ�����HbLeproe������»���ȱʧ7kb���¦Ħ��ںϻ����γɦ�0��ƶ��
��������ȱ�ݲ�
����ȱ�ݲ���immunodeficiency����ָ����ϵͳ�����ϰ������һ�༲��������ԭ��ɷ�Ϊ�Ŵ��ԣ�ԭ���ԣ���̷��������ࡣ���ݲ������߷�Ӧ��ϸ���ֿɷ�ΪBϸ����Tϸ���Լ���������ȱ�ݲ���������Ҫ�����������߹��ܵĸ��ֵ������Ŵ���ȱ�����������ȱ�ݲ�����Ҫ�����м��֣�
1.�ޱ���Ѫ֢ �ޱ���Ѫ֢��agammaglobulinemia����Ϊ���ͣ�
��1��Bruton�ͣ����ͳ�X���������Ŵ���Ѫ��Bϸ��ȱ�磬����Ѫ��IgA��IgG��IgM��ȫ�����ȱ�硣��Tϸ������Ӱ�졣���������������ص�ϸ����Ⱦ�����Բ���������������ԣ�ϸ���������б��棬����ֲ�����ųⷴӦ������һ��X�����ĵͱ���Ѫ֢��hypogammaglobulinemia�����ߣ�������Ⱦ�⣬�а�����������ȱ������������������ٻ������İ�С���Է����ӳٵ�֢״��Fleischer �ۺ�������
��2����ʿ�ͣ���ʿ�ͣ�Swiss type���ʳ�Ⱦɫ�������Ŵ���Ѫ��Bϸ����Tϸ����ȱ�磬���ٷ�����ȫ����ϸ��������������ֿ������ͣ�����ֲ�����ų�������Ѫ��IgA��IgG��IgM��ȱ������ص��¡�Ԥ���Bruton�������1���������������������ԣ�Ҳ������X�����ʹ��ڡ�
2��������������ȱ�ݲ� ������������ȱ�ݲ���severecombined immunodeficiency����ָBϸ����Tϸ������ȱ�ݵ�һ������ȱ�ݲ���һ���Ϊ4�࣬��ʿ���ޱ���Ѫ֢������һ�࣬����3��Ϊ��X�����ܰ�ϸ�������Եͱ���Ѫ֢����״ϸ��������ȫ�������Ѱ�øȱ��֢��
3��ѡ����������ȱ��֢ ���ڼ�����������ĸĽ����������п��ܽ��ͱ���Ѫ֢������Ҫȱ�������������ͷ�Ϊ8�ࣨ��4-2����
��4-2ѡ�������ߵ���ȱ��֢����
| IgAȱ��֢ ѡ����IgAȱ��֢ ����ʧ��-ëϸѪ������֢ Nezelof�ۺ�֢ ����Ƥ��ճĤ������� SIgA�������ͣ�ȱ��֢ IgAȱ��֢ IgAȱ��֢ ѡ����IgMȱ��֢ Wiskott-Aldrich�ۺ��� IgEȱ��֢ IgA-IgMȱ��֢ IgA-IgGȱ��֢ L��ȱ��֢ |
ѡ����IgAȱ��֢Ϊ������͡�������Ҫ����Ϊ�Ϻ�������Ⱦ�����ס�֧�����ף����Ϊ���ס����������Լ��������ʪ�ؽ��ס�ϵͳ�Ժ���Ǵ����ȡ�
4������ɷ�ȱ��֢ ���壨cpmplement����һ���Ƚϸ��ӵ�ϵͳ������12�ֳɷݣ����нӽ��������ʽ�����ϵͳP���ӣ������أ���B���Ӻ�D���ӣ����⣬���м��ֵ��ڵ���B��Ci�������ӣ�Ci INH����C3b��������(3bINA)����IH���ӵȡ�
����ͻ����Ե��²���ϳ��ϰ���������ȱ��֢���ѱ�����IJ���ȱ��֢������14�֣�������Ci INH,C1q,C1r,C2,C3,C3b������C4��C5��C6��C7��C8��C9ȱ��֢��C5�����쳣�����༲������Ҫ�����Ƿ�����Ⱦ��Ƥ�º�ճĤˮ�����������Լ�����
����Ĥ���ײ�
ϸ��Ĥ�����ʡ���������Ϣ�ܵĴ��ݺͱ任�ij��������й㷺���������ܣ����������������Ĥ����ϢϢ��صġ����书�ܿɷ�Ϊ���м��ࡣ
1��Ĥ�Ǽܵ��ײ� �Ժ�ϸ��Ϊ������ϸ��Ĥ����˫��֬����ɣ������ҪΪ������֬���ڲ���ҪΪ������֬�����˫֬�㼰˫֬�����ж��ֵ�����*�����˫֬��ĵ����ʳ�Ϊ����Ĥ�����Ĥ���ף�transmembrane proteins��(ͼ4-17)�������У���Ѫ���ǵ���A��B��C��glycophorin,GP-A,B��C��,�п�ԭ�����幦�ܣ��ڵ���3Ϊ��Ҫ��������ת�˵��ף���Ϊ����������
* Ĥ�����������õ�Ӿ���ʿ�������Ϊ����1-8�������ַ�Ϊ�Ǵ����ף��絰��4�е���4.1��4.9�ȣ�����Щ�����ʵ����ʻ�����ȷ���е��������иı䣬�絰��2.1�ֳ�Ϊê���ȡ�
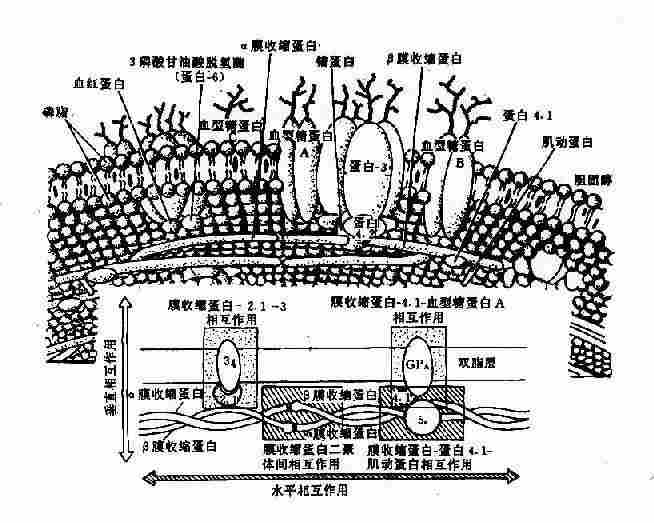
ͼ4-17 ��ϸ��Ĥ�Ľṹʾ��ͼ
֬���ڲ�����ʽӴ����ݵĵ����ʳ�����Ĥ���ף�peripheral membrane protein���������У���Ĥ�������ף�spectrin,Sp������Ҫ�ɷ֣�������������ɣ�����-Sp(240kd)���-Sp(220kd)�γɵĶ����壨�쳣�����壬heterodimer������Ĥ�����ף�actin,����5�����ʶ�˿״������Sp���ľ��壻�۵���4.1�书��Ϊ�ٽ�Sp��Ĥ������ϣ�����GP-C���ӣ��༴��Sp�̶���Ĥ�ϣ��ܵ���4.2������������ͨ����ê��������4.1��ϣ��ݵ���4.9����Sp-Sp�ľ����ϣ�����Sp�͵���4.1��ϣ����ɺ�ϸ��Ĥ�Ǽܣ�membrane skeleton�������壨ͼ4-17������ê���ף�ankyrin,����2.1�����������ƺ������Ӧ�-Sp�뵰��3��ʹĤ�Ǽܵ��̶���֬���ڲ㣬��ê�������á���������һЩĤ���ף���������̽���С�Ĥ�Ǽܵ��γ���ά�ֺ�ϸ��˫���νṹ��Ĥ�Ŀɱ����Ժ������ԵĻ���������ͻ�䵼����ЩĤ���ṹ���ܵĸı䣬��һ���Ŵ�����Ѫ��ƶѪ�ĸ���ԭ��
��1������ϸ������ƽ������ϸ������֢��spherocytosis���������κ�ϸ������Ϊ�ص����Ѫ�Լ��������߳������ж�ƶѪ�����㡢Ƣ�������ϸ�������Σ������������ߡ����������ʳ�Ⱦɫ�������Ŵ����ӷ���ˮƽ�������Ŵ������ԣ���Sp�����ж�ȱ����ê����ȱ��������4.2ȱ���Ȳ���
��2����Բ��ϸ������֢����������Ѫ����1%-14%����Բ�κ�ϸ�������Ŵ�����Բ�κ�ϸ������֢ʱ��������50%-90%��������Ѫ��ƶѪ�����㡢Ƣ���֢״����ϸ���������ߡ���ʳ�Ⱦɫ�岻��ȫ�����Ŵ������Ŵ�����Ҳ�������ԣ�����Sp���������ϰ�������4.1�쳣���ǵ���ȱ������
��3��������ϸ������֢��������ϸ������֢��pyropoikilocytosis,HPP�����ص�������Ѫ��ϸ�����Ȳ��ȶ����ڼ�����46��ʱ���������ϸ��������ϸ����Ƭ������Ҫ49��ų��֣������߱���������Ѫ���ʳ�Ⱦɫ�������Ŵ���Sp����ȱ���DZ����Ļ���ԭ��
Ŀǰ���ֵļ�ʮ��Ĥ������ͻ�������������йأ���ij��ͻ���������ּ����䲢û�з��ֱ�Ȼ����ϵ����Spͻ����ռ������
2��Ĥת�˵��ײ� ��Щ���ʣ��簱���ᡢ�����ǣ�ͨ��ϸ��Ĥ��Ҫ�����ڷ��ӵ��壨Ĥ���ף���������ɢ��faciliteddiffusion��������ͻ��ɵ���Ĥ�����ʻ����ĸı䣬Ӱ��ijЩ����ͨ��ϸ��Ĥ��ת�ˣ��ɴ˲�����һ�༲����Ϊת�˲���transport disease���������װ�����֢��������������С�ܺ�θ����Ƥϸ������ת���ס�������������4�ְ����ᡣ�����ر����װ����ȵͣ�������Ũ�ȳ���30mg%ʱ�����ײ����װ����ʯ����ˣ���·��ʯ����Ⱦ����ʹ�DZ�������Ҫ�ٴ����֡�Ŀǰ��֪��ת�˲���ʮ���֡�
3����Ĥ���ײ� ��Ĥ���ײ��Ը����ڼ�Ĥ��sarcolemma���ϵĿ���ή�����ף�dystrophin,�ֳƼ�Ӫ���������ף��Ŵ�ȱ��Ϊ�����ļٷʴ��ͼ�Ӫ������֢���ֳ�Duchenne�ͼ�Ӫ������֢��DMD���ͱ��ֽ��۵����Լٷʴ��ͼ�Ӫ������֢���ֳ�Becker�ͼ�Ӫ������֢��BMD���������ۡ�
DMD��һ�������²�������X���������Ŵ�����������ԼΪ1/3500����Ӥ�����ٴ������Լ���Ľ�����ή��������Ϊ���������߶���3-5�귢����������������·���ѳ�Ѽ�������ֻ������賦�����Էʴ�����Ͳ�ͬ�̶ȵ��ļ���Լ30%���������ϰ�������άή�����ԣ���֬���������֯�����Ѫ�����ἡ�ἤø�Ȼ������ߡ�����12�����ұ㲻�����ߣ�һ����20��ǰ������BMD���߲�����ᣬ�ٴ�������DMD���ƣ���5-25�귢�������̷�չ�ϻ���������ڽϳ����л���63���ߡ�������֤ʵDMD��BMD��ͬһ��������ͬͻ��ĺ����
��Ⱥ��DMD����Լ2/3�м���ʷ����X���������Ŵ�����Լ1/3����Ϊɢ�������ɻ�����ͻ����ɡ�DMD����λ��Xp21��ȫ��Լ2300kb��Ϊ����������ʶ�������˻������ٺ���79�������ӣ��ں��ӳ��Ȳ���ܴɴ�1kb-180kb��cDNAȫ��13,974bp������3685�������ᣬ������Ϊ427kd�����ݵ�����ʾ�û������Ϊһ��״�ṹ��ϸ���Ǽܵ��ף��ɷ�Ϊ4����������ͼ4-18����
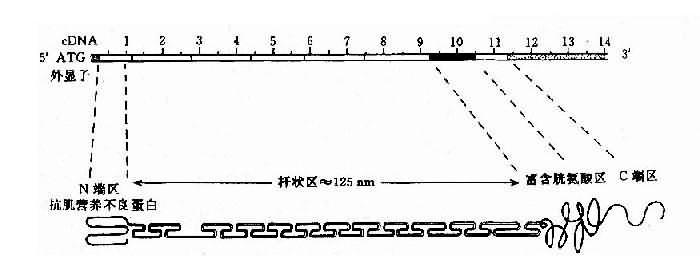
ͼ4-18 DMD��������뵰���ʵĽṹʾ��ͼ
Ŀǰ��֪��DMD����ͻ����ҪΪȱʧ�ͣ�Լռ������50%-60%���ظ�(duplication)��֮��Լռ6%��������ȱʧ��������5���˵ĵ�4-21�����ӣ�ռȱʧ��20%������һΪ��45-52�����ӣ�ռ54%-60%�����ں���44Լ160-180kb������Ƶ����ߣ�ȱʧ��������ͻ���ߣ�����������DMD��δ���������߶�ΪBMD������ȱʧ���ظ������п���ʹͻ������������ж��γ�����Ƭ�Σ�����ڼ�ϵ������Я���������ǰ������϶��������塣
DMD�����ϻ�����һЩ�̴����ظ�˳��short tandem repeats,STR������֪��13��STR��DMD��STR�����Ϊ�����������ǵ�Ԫ�Ĵ����ظ�����CA������Ca repeats�����ظ�Ƶ���ڲ�ͬ�������ܴ��Ӻ���Ƶ�ʴ�25%-93%��Ϊ��ȱʧ��DMD�IJ�ǰ��ϡ�Я������ṩ��һ����Ч���Ŵ���ǡ�
Ŀǰ������Ч�Ʒ���Ψһ��Ч��Ԥ��;���ǶԸ߷���̥�����в�ǰ��ϣ����ĵ�ʮ���£���
�ġ���Ѫ������Ѫ����ȱ��֢
��Ѫ��һ���ӵ��������̣�����������Ѫ���Ӽ�����Ѫ���Ӳ��롣�Ŵ�����Ѫ�ϰ����������������������ǰ�������ڻ���ͻ������Ѫ���ӻ��Խ��ͣ����������ڿ���Ѫ���ӻ���ȱ����
1����Ѫ����ȱ��֢
��Ca2+����Ѫ�����⣬������Ѫ���Ӷ����Ŵ���ȱ���ı��棬����Ѫ�Ѳ����ر��Ǽ���Ѫ�Ѳ���Ϊ���������Ҫ��Ѫ�Ѳ��������йص�Ѫ���Լ�Ѫ�Ѳ����Խ��ܡ�
Ѫ�Ѳ���hemophilia�����ס��ҡ��������⣬���Ϻ����ַ��ֵ�һ��vWF���ӣ�vonwillibrand factor��ȱ����Ѫ���Լ�Ѫ�Ѳ�������Ѫ�Ѳ����������͡�1986-1989��ȫ��24ʡ���У�37��������Ѫ�Ѳ������ʵĵ��飬��������16866 654�ˣ��ܻ�����2.73/10��(����5.21/10��Ů��0.06/10��)����ŷ����Ƚϵͣ����͵Ĺ��ɱ��ǣ�����79.8%������14.1%������2.8%��Ѫ���Լ�Ѫ�Ѳ�3.3%��
��1������Ѫ�Ѳ�������Ѫ�Ѳ���hemophilia A��������Ѫ�Ѳ��ף�antihemophilicglobulin,AHG��ȱ��֢��ڢ�����ȱ��֢����Ҫ����Ϊ��Ѫ�������Ѫ�ص�Ϊ���ٻ���������Ѫ���ڶ����������֮�۳�Ѫ��λ�㷺�����������������γ�Ѫ�ף����ڱ��Σ������Ϊ�ڳ�Ѫ��
��֪�������������ֳɷ���ɣ���F����C��AHG������F����Ag(��������ؿ�ԭ)����vWF���ӡ�����Ѫ�Ѳ�ΪAHG�Ŵ���ȱ�����¡�
����ΪX���������Ŵ�������λ��Xq28,�����ȳ���186kb����26��������(ռ9kb)��25���ں���(ռ177kb)��ɣ�����2351�������ᣬ�ѷ���ȱʧ�ͣ��������塢���弰����ͻ��ȣ���46�����ϡ��Ӻ��ӵļ����Կ�չ�Ŵ���ѯ����Ҫ����ȥ��ͨ���ⶨѪ��AHGˮƽ���â�R��Ag/AHG��ֵ������Ӻ��ӣ������ܲ�ȡ�����Ŵ�ѧ�ֶΣ��ر����ѳɹ���Ӧ��DNAӡ���ӽ���PCR�������ڲ�ǰ��ϣ���Է�ֹ���ͻ���������ʮ����Ч��
���ƿ�ʹ�ø���AHG�Ƽ������賤��ʹ�ã������о��Ļ������ƽ����DZ����ĸ��η�����
��2������Ѫ�Ѳ�������Ѫ�Ѳ���hemophilia B������Ѫ����Ѫ��ø�ɷ֣�PTC��ȱ��֢��ڢ�����ȱ��֢�������ٴ����ֿ��Ƽ��ͣ��������ʽϵͣ��Ŵ���ʽ��Ϊ�����������Ŵ��������Ӻ��Ӣ����ӻ��Խ�Ϊ����1/3��ijЩ�Ӻ��ӿɳ���֢״����Ů�Բ��˽ϼ��Ͷ����
����ڢ����ӻ����Ѷ�λXq27.1�������ܳ���Ϊ34kb���ң���8����������ɡ��Ѽ�������ͻ����100��֮�ࣨ����ȱʧ��ȫȱʧ��30�֣�����Ϊ�������͵�ͻ�䣩���ҹ��������ȱ��������췢�ֵ�5�ֵ�ͻ�䣨������2��5�����Ϻ������ϵȷ���1��1-3�ں���5kbƬ��ȱʧ������Щͻ���з��ּ���������ͻ�䣬�ٴ�����Ϊ��ͯ�������س�Ѫ�������ഺ�ں��Է���Ѫ���ᡣ��֤����������̴����յ������Ӳ��������ӣ���Ҳ�����ٴ�֢״������ͻ�����ʿ�����һ����ϵ��
������������Ѫ����Ũ��Ѫ���Ƽ����ƣ����ڢ����ӻ�����ߵ�25%���ϼ�����Ч����ǰ����Ƿ�ֹ����������������Ч���������꣬�ҹ������ڢ�����ȱ��֢�Ļ������Ʒ���ȡ���˽�չ���������ٴ�ȡ�ó����ȶ���Ч��
��3������Ѫ�Ѳ�������Ѫ�Ѳ���hemophilia C������Ѫ����Ѫ��øǰ�ʣ�PTA��ȱ��֢��plasma thromboplastic antecedent deficiency����ڢ�����ȱ��֢������֢״�ϼס������ᡣ���������������ԣ�������������ϲ���̫�˺��ᡣ�Ŵ���ʽ��Ⱦɫ�������Ŵ�����֪������λ��15q11������Ϊ23kb����15����������ɣ�����625�������ᣬ������11-15�����ӱ����Ȼ��˾�����Ѫ����Ϊ��������Ҫ�ɷ֡��ѷ���3�ֵ�ͻ�䡣���������ӵĢ����ӻ�����20%���£��Ӻ���Ϊ30%-65%����������ȱ������С����������Ũ��Ѫ�����Ƽ���Ч��
��4��Ѫ���Լ�Ѫ�Ѳ���Ѫ���Լ�Ѫ�Ѳ���von Willebrand disease����һ�ֽ϶������ڢ������йص��Ŵ�����Ѫ�ϰ����뱾���йص�von Willebrand���ӣ�v WF����һ����������ǵ��ס�����λ��12pter-p12,vWF����Ϊ180kb����52�������ӣ�mRNA�ܳ���Ϊ9kb���ұ���2813�������ᡣvWF������Ѫ����Ƥϸ��������ϸ�����ڣ�����Ѫ�в�����Ϊ���������壬���ҿ���ǿ���ȶ��ԣ���vWF����ȱ���������Т����ӻ��Խ��͡����⣬ѪС��������к�vWF����Ҳ����ѪС��ۼ�������Ѫ�з������á��������������Եij�Ѫ����Ѫ��AGH���Խ��ͣ����������Ѫ�Ѳ����أ��ӻ���ˮƽ�ѷ���20����ͻ���͡�
���ڶԱ����������㹻���˽⣬�ѿ�ͨ��RFLP����������PCR���Ա������в�ǰ��ϡ�
���⣬�����Ŵ���ѪС��ȱ�������ϰ�����ά����ԭ���Ŵ���ȱ�ݶ�����ɵ���״̬���ڴ˲�������
2������Ѫ����ȱ��֢
��1���Ŵ��Կ���Ѫø��ȱ��֢������Ѫø��antithrombin��,AT����ѪøXa���������ã������ܼ��������Ѫø�����ơ���Σ�AT�������Ƣ����������Ĺ��ܡ�
�Ŵ��Կ���Ѫø��ȱ��֢��hereditary antithrombin �� deficiency�����ٴ�����Ϊ������Ѫ˨�γɣ���Ҫ��λΪ�ľ���������������
����Ϊ��Ⱦɫ�������Ŵ����������ڲ�ͬ�������������졣ŷ���������пɸ�1��2000-5000���ҹ�Ҳ���и������档��֪��AT�����λ1q23������16kb����7����������ɣ�����432�������ᣬ�����ѷ���20�����ϵ�ͻ�����ͣ����ֳ���ͬ�Ĺ���ȱ�ݡ�
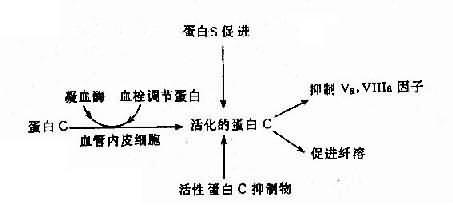
��2���Ŵ��Ե���Cϵͳ�쳣���Ŵ��Ե���Cϵͳ�ɵ���C��PC��������S��PS����Ѫ˨���ڵ��ף�thrombomdorlin,TM�������Ե���C�����APC1����ɣ�����֮��Ĺ�ϵ��ͼ4-19����ĵ���C������Va����a���ٽ���ά�����ܽ�����á�����Cϵͳȱ��֢��3�֣�
ͼ4-19 ����C������S�����Ե���C����������ϵ
1������Cȱ��֢����Ҫ֢״�Ǿ���Ѫ˨�γɣ�����������������������ʹ���Ϊ1��16000���ʳ�Ⱦɫ�����ԣ�����ȫ���ԣ��Ŵ������������أ�����Ϊ��Ѫ��Ƥ��������������Ѫ������Ѫ��DIC����Ѫ˨���Ӻ��Ӷ�������׳�귢��������C����λ2��Ⱦɫ�壬����12kb����8����������ɣ��Ѽ�����������ȱʧ�ͼ���ͻ�䲡����Ҳ���漰mRAN�ӹ�ȱ���ߡ�
2������Sȱ��֢������S�������Ǵٽ������C��APC���������֬������APC���Va���ӡ�����������Ⱦɫ�����ԣ�����ȫ���ԣ��Ŵ�������λ��3��Ⱦɫ�壬���ܳ���Ϊ45kb���Ϻ�Ҳ�ѱ���2��PSȱ��֢��
3�������Ի����C����ȱ��֢��congenitaldeficiency of activated protein Cinhibitor����
���������Ŵ�������ϵͳ�쳣���������������쳣����øԭѪ֢��������������øԭ�������ͷ��쳣�����Ŵ�������øԭ��������������֢����Ѫ���쳣�������������ܼ����ȣ������ֳ�����״̬�ĸ���֢״��
�塢���嵰�ײ�
�����Ǵ�����ϸ��Ĥ�ϡ������л���ڵ�һ���������ܵĵ����ʡ�����֤�����е����������ܺ������������30�����ϡ����а������ļ������塢�̴��༤�������Լ����ʡ�ǰ�����ء����������ӡ�֬�������塣��������ı����ǵ����ʣ����Զ���������ͻ��Ҳ�ɵ������嵰���ʺ����ĸı䡣�������嵰���Ŵ���ȱ�������һ�༲������Ϊ���岡��receptor diseae����
���岡�л�������Ŵ���֮�֡��Ŵ������岡�о��ý϶��������4�֣�
1�������Ըߵ��̴�Ѫ֢ �����Ըߵ��̴�Ѫ֢��familialhypercholesterolemia,FH�����ٴ������ǣ�����ʱ�����ڸߵ��̴�Ѫ֢�����ߵĵ��̴���ҪΪ���ܶ�֬�����̴�����LDL-C���ͦ¼����ܶ�֬���ף���-LDLC������ɫ��(xanthoma)�����ߵĵ��̴�����֯�㷺�������������صĺ�������귢����������Ӳ������������5-30�꼴�����Ľ�ʹ���ļ�����֢״�����������Ӻ��ӷ������IJ��Գ��ҷ����ʽϵ͡�
����Ϊ��Ⱦɫ�������Ŵ���������Ⱥ�У��Ӻ��ӷ�����Ϊ1��500���Ӻ����ٴ����ֽ��ᣬ��������ȫ�����Ŵ��������ʸߣ�90%-100%����
��������Ҫ������LDL���壨LDLR�����Ŵ���ȱ����Ѫ���е�LDL�ͦ�-VLDL��LDL�����Ϻ�������ϸ��������ø����LDL��LDLR���룬LDLR�ص�ϸ�������������ã�LDL���ڵ��̴���ø�����£��ͳ����̴���ϸ�����á�����һ�����У�ϸ�������й��ܵ�LDLR����������Ѫ����LDL����-VLDL��Ũ�ȡ���֪LDLR�ı������λ��19p1.3-p13.3���ܳ���45kb����18����������ɡ��ѷ���FH��������ʮ��ͻ�䣬����ȱʧ��ͻ�䣨��Ҫ�ģ�������ͻ�䡢����ͻ�䡢����ͻ�估����ͻ�䡣����ΪLDLR�����쳣��4�����ͣ���ϸ��Ĥ����ȫ��LDLR����LDLR���Լ��٣���LDLR���ܲ�����LDL��ϣ���LDLR��LDL��Ϻ������̣����������ںϳ�����ǰ���мӹ�ȱ�ݡ�
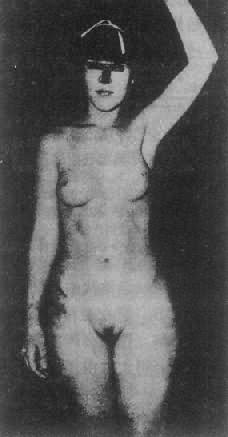
2��غ��Ů�Ի��ۺ��� غ��Ů�Ի��ۺ�����testicularferminization syndrome���ֳ�������ȫ�������ۺ�����complete androgeninsensitivity syndrome,CAIS��,Ϊ������Լ����Ի��Ρ����ߺ���Ϊ46��XY����غ�裬�ܷ��������أ���ȱ�����������壬�����ز��ܷ���ЧӦ��ͼ4-20����
ͼ4-20غ��Ů�Ի��ۺ������ߣ�����46��XY��