第二节 常用抗高血压药
一、主要影响血容量的抗高血压药
利尿药是治疗高血压的常用药,要单独治疗轻度高血压,也常与其他降压药合用以治疗中、重度高血压。一般认为利尿药初期降压机制是排钠利尿、造成体内Na+、水负平衡,使细胞外液和血容量减少之故。长期应用利尿药,当血容量及心输出量已逐渐恢复至正常时,血压仍可持续降低,其可能机制如下:①因排钠而降低动脉壁细胞内Na+的含量,并通过Na+-Ca2+交换机制,使胞内Ca2+量减少。②降低血管平滑肌对血管收缩剂如去甲肾上腺素的反应性。③诱导动脉壁产生扩血管物质,如激肽,前列腺素等。
摄入大量NaCl能对抗利尿药的降压作用,限制NaCl摄入则能增强其降压作用,说明排Na+是利尿药降压的重要原因。
临床治疗高血压以噻嗪类利尿药为主,但长期应用常致不良反应如:降低血钾、钠、镁,增加血中总胆固醇,甘油三酯及低密度脂蛋白胆固醇含量,增加尿酸及血浆肾素活性。大剂量噻嗪类利尿药还可加剧脂血症,降低糖耐量等。但使用低剂量的双氢氯噻嗪,则可避免代谢方面的副作用,其他如呋噻米、氨苯蝶啶等也可应用。
一般情况下,高效利尿药不作为轻症高血压的一线药,而用于高血压危象及伴有慢性肾功能不良的高血压患者,因其不降低肾血流,并有较强的利Na+作用。
二、β受体阻断药
β受体阻断药均有良好的抗高血压作用,现以普萘洛尔为例介绍如下:
【抗高血压作用】用普萘洛尔数天后,收缩压可下降15%~20%,舒张压下降10%~15%,合用利尿药降压作用更显着。静脉注射普萘洛尔后可使心率减慢,心输出量减少,但血压仅略降或不降,这是压力感受器反射使外周阻力增高的结果。
有少数人,使用β阻断药后,总外周阻力增高,推测是激活了血管的α受体,故外周血管有病者,禁用本药。
【作用机制】普萘洛尔降低血压是其β受体阻断作用所继发的,对其进一步机制有以下几种看法。
1.减少心输出量 普萘洛尔阻断心β1受体,抑制心肌收缩性并减慢心率,使心输出量减少,因而降低血压。给药后这一作用出现迅速,而降压作用出现较慢。
2.抑制肾素分泌 肾交感神经通过β1受体促使邻球器分泌并释放肾素,普萘洛尔能抑制之,从而降低血压。具有强内在活性的吲哚洛尔在降压时,并不影响血浆肾素活性。
3.降低外周交感神经活性 普萘洛尔也能阻断某些支配血管的去甲肾上腺素能神经突触前膜的β2受体,抑制其正反馈作用而减少去甲肾上腺素的释放。
4.中枢降压作用 已知下丘脑、延髓等部位有β受体,中枢给予微量普萘洛尔能降低血压,同量静脉注射却无效。与之相反的证据是,不能进入中枢的β阻断药,却有降压作用。因此中枢β受体在血压调节中的意义,尚待阐明。
β受体阻断药的作用机制较为复杂,可能在某种病情发展中某一机制起主导作用,而在另种病情过程中,另一机制占主要地位。
【临床应用】β受体阻断药已广泛用于治疗高血压,对轻、中度高血压有效,对高血压伴心绞痛者还可减少发作。此外,对伴有心输出量及肾素活性偏高者,对伴脑血管病变者疗效也较好。普萘洛尔的用量个体差异较大,一般宜从小量开始,以后逐渐递增,但每日用量以不超过300mg为宜。在β受体阻断药中,选择性β1受体阻断药美托洛尔(metoprolol),阿替洛尔(atenolol)的作用优于普萘洛尔,它们在低剂量时主要作用于心脏,而对支气管的影响小,对伴有阻塞性肺疾患者相对安全些。
三、钙拮抗药
钙拮抗药能抑制细胞外Ca2+的内流,能松弛平滑肌、舒张血管,使血压下降。降血压时并不降低重要器官的血流量,不引起脂质代谢及葡萄糖耐受性的改变(详见二十一章)。
硝苯地平
硝苯地平(nifedipine)对轻、中、重度高血压者均有降压作用,但对正常血压者则无降压效。口服30~60分钟见效,持效3小时,t1/2约3~4小时。在离体血管实验中,它能明显抑制高钾去极化所致的收缩反应,对去甲肾上腺素所致的收缩反应则抑制较弱,但对自发性高血压大鼠的血管标本,由去甲肾上腺素所引起的收缩反应却有明显的抑制作用,这似能说明硝苯地平对血压正常者无降压作用的理由,此外,也可抑制内皮素诱导的肾血管的收缩。
硝苯地平降压时伴有反射性心率加快和心搏出量增加,也增高血浆肾素活性,合用β受体阻断药可免此反应而增其降压作用。
临床用于治疗轻、中、重度高血压,可单用或与利尿药、β阻断药合用。
常见不良反应有头痛、脸部潮红、眩晕、心悸、踝部水肿等。其引起踝部水肿为毛细血管前血管扩张而不是水钠潴留所致。
同类药物维拉帕米、地尔硫
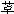
粉防已碱
粉防已碱(tetrandrine)是中药粉防已根中所含的生物碱,对自发性高血压大鼠及高血压患者均有明显的降压作用,为其钙拮抗作用所致,已证明它能抑制T及L型钙通道。一般口服给药,对重度高血压及高血压危象者可静脉注射。
本药无严重不良反应,少数患者有轻度恶心及上腹部不适等。
四、影响血管紧张素Ⅱ形成的抗高血压药――血管紧张素Ⅰ转化酶抑制剂
肾素-血管紧张素-醛固酮系统(RAAS)在血压调节及高血压发病中都有重要影响。
近几年来合成了一系列血管紧张素转化酶抑制剂(angiotensinconverting enzyme inhibitors,ACEI),如卡托普利(captopril),依那普利(enalapril),雷米普利(ramipril),赖诺普利(lysinopril)及培哚普利(perindopril)等。它们能有效地降低血压,对心功能不全及缺血性心脏病等也有良效。
现代分子生物学研究证明,在心血管、脑、肾等组织中存在着肾素、血管紧张素原的mRNA,局部有相关基因表达,故提出在组织中存在独立的RAAS(系由局部合成),该系统以旁分泌及自分泌方式对心血管及神经系统功能,甚至结构起调节作用。血管中局部产生的血管紧张素Ⅱ(ATⅡ)可增加血管的收缩性能,并促进去甲肾上腺素的释放,而导致血管收缩,血压上升,实验见ATⅡ能促进培养的血管平滑肌细胞生长、增殖,增加蛋白质合成及细胞体积。
ATⅡ促进血管平滑肌生长的作用可引发血管增生及血管壁中层增厚等。
【药理作用及作用机制】ACEI能使血管舒张,血压下降,其作用机制如下:
1.抑制循环中RAAS
ACEI主要通过抑制ATⅡ的形成而起作用,对血管、肾有直接影响。并通过并感神经系统及醛固酮分泌而发生间接作用。这是改变血流动力学的主要因素,也是用药初期外周血管阻力降低的原因。
2.抑制局部组织中RAAS
组织RAAS对心血管系统的稳定有重要作用,组织中的血管紧张素Ⅰ转化酶(ACE)与药物的结合较持久,因此对酶的抑制时间更长,进而降低去甲肾上腺素释放,降低交感神经对心血管系统的作用,有助于降压和改善心功能。此与药物的长期降压疗效有关,药物与ACE的结合方式见图26-1,以卡托普利为例,卡托普利的三个基团可与酶的三个活性部位相结合,一是脯氨酸羧基与酶的正电荷部位(精氨酸)呈离子键结合;二是肽链的羰基与酶的供氢部位呈氢键结合;三是巯基与酶的Zn2+结合,终使酶失去活性。
3.减少缓激肽的降解 当ACE(即激肽酶Ⅱ)受到药物抑制时,组织内缓激肽(bradykinin,BK)降解减少,局部血管BK浓度增高。BK是血管内皮L-精氨酸-NO途径的重要激活剂,它作用于内皮的β2-受体而引起EDHF(血管内皮超极化因子)及NO的释放,因而发挥强有力的扩血管效应及抑制血小板功能。此外,BK可刺激细胞膜磷脂游离出花生四烯酸(AA),促进前列腺素的合成而增加扩血管效应,见图26-2。
图26-1 卡托普利与酶的活性部位结合图
图26-2 ACEI对RAAS及激肽释放酶-激肽-前列腺素系统的影响
ACEI与其他降压药相比,具有以下特点:
1.适用于各型高血压,在降压的同时,不伴有反射性心率加快,可能是取消了ATⅡ对交感神经传递的易化作用所致。
2.长期应用,不易引起电解质紊乱和脂质代谢障碍,可降低糖尿病、肾病和其他肾实质性损害患者肾小球损伤的可能性,如卡托普利既能有效降压,又能增加机体对胰岛素的敏感性。
3.可防止的和逆转高血压患者血管壁的增厚和心肌细胞增生肥大,可发挥直接及间接的心脏保护作用。
4.能改善高血压患者的生活质量,降低死亡率。
【体内过程】见表26-1
表26-1
| 参数 | 卡托普利 | 依那普利 |
| 前体药 | 否 | 是 |
| 活性代谢产物 | / | 依那普利拉 |
| 生物利用度(%) | 70 | 40 |
| 血浆蛋白结合率(%) | 30 | 50 |
| t1/2(h) | 2 | 30 |
| 维持时间(h) | 3~4 | 12~24 |
| 消除途径 | 肾 | 肾 |
| 12.5-50mg | 10-40 | |
| 剂量 | 2~3次/日 | 1~2次/日 |
【临床应用】治疗原发性及肾性高血压能使血压降低15%~25%,对中、重度高血压合用利尿药、可加强降压效、降低不良反应。
【不良反应】虽不良反应发生率较低,但也不是绝对安全的,主要不良反应有低血压(2%),见于开始剂量过大时,应小量开始试用。高血钾、血管神经性水肿。肾功能受损,对肾血管狭窄者更甚。咳嗽,为刺激性干咳,可能与肺血管床内的激肽及前列腺素等物质的聚积有关。久用可致血锌降低而引起皮疹、味觉、嗅觉缺损、脱发等。补充Zn2+可望克服。
【药物相互作用】合用利尿药可增强降压效,并减少Zn2+的排泄;吲哚美辛可减弱卡托普利的降压效,此与吲哚美辛抑制前列腺素的合成有关;与地高辛合用,可增高地高辛的血浆浓度等。
五、交感神经抑制药
(一)主要作用于中枢部位的抗高血压药
可乐定
可乐定(clonidine,可乐宁)为咪唑类衍化物。
【药理作用】麻醉动物静脉注射可乐定后,先见血压短暂升高,随见血压持久下降,伴有心率减慢、心输出量减少。升压是可乐定激动外周血管α受体所致,随后的降压则与中枢作用有关。口服给药仅见降压而无升压效,继续服用后,外周血管阻力逐渐降低,肾血管阻力也降低,但并不显着影响肾血流量及肾小球滤过率。
可乐定的降压作用中等偏强。它还能抑制胃肠道的分泌和运动,因此适用于兼患溃疡病的高血压患者。
可乐定对中枢神经系统还有镇静作用,减少自发性活动,并显著延长巴比妥类的催眠时间。
【作用机制】动物实验证明微量可乐定注入椎动脉或小脑延髓池均可引起降压,但同等量作静脉注射却并不降压,据此推测,引起降压作用的部位在中枢。通过分层切除脑组织,提示可乐定作用于延髓并降低外周交感神经功能而降压。晚近已证明可乐定引起血压下降的机制是激动了延髓腹外侧嘴部(rostralportion of the ventrolateral medulla)的Ⅰ1-咪唑啉受体(Ⅰ1-imidazoline receptor),降低外周交感张力致血压下降。而其激动中枢α2受体则是其引起镇静等副作用的原因。
此外,从动物脑中已提得内源性可乐定样物质,该物质作用于延髓腹外侧发挥类似可乐定样的作用。另有研究证明,可乐定降压涉及到内源性阿片肽的释放。且可乐定具有镇痛效,此效可被阿片拮抗剂-纳洛酮所拮抗。可乐定也激动外周交感神经突触前膜的α2受体及其相邻的咪唑啉受体,引起负反馈而减少去甲肾上腺素的释放。可见其降压机制复杂。
【体内过程】可乐定口服吸收良好,生物利用度约75%,口服半小时后起效,2~4小时作用达高峰,持续6~8小时。在体内分布均匀,也易透过血脑屏障。t1/2为7.4~13小时。约50%在肝代谢,使结构中的咪唑环裂解,苯环被羟化。余者以原形随尿排出。
【临床应用】可乐定可治疗中度高血压,常于其他药无效时应用。此外,可作为吗啡类镇痛药成瘾者的戒毒药。
【不良反应】常见不良反应有口干,为其作用于胆碱能神经末梢上的α2受体,减少Ach的释放和过量唾液的分泌所致。久用使水、钠潴留,是降压后肾小球滤过率减少的结果。合用利尿药可克服。此外还有镇静、嗜睡、头痛、便秘、腮腺痛、阳萎等,停药后能自行消失,少数患者在突然停药后可出现短时的交感神经功能亢进现象。如心悸、出汗、血压突然升高等,可能是久用后突触前膜α2受体敏感性降低,负反馈减弱,去甲肾上腺素释放过多所致。再用可乐定或用α受体阻断药酚妥拉明能取消之。
甲基多巴
甲基多巴(methyldopa)的降压作用与可乐定相似,属中等偏强,降压时也伴有心率减慢,心输出量减少,外周血管阻力明显降低。治疗中度高血压,适用于肾功能不良的高血压患者。
(二)抗去甲肾上腺素能神经末梢药
利血平是印度萝芙木所含的一种生物碱,国产萝芙木所含总生物碱的制剂称降压灵。该药降压作用弱,不良反应较多,现已少用。作用较强的胍乙啶也因不良反应多而少用。
(三)肾上腺素受体阻断药
α受体阻断药
哌唑嗪
哌唑嗪(prazosin)是人工合成的喹唑啉类衍生物。
【药理作用】哌唑嗪能选择性地阻断突触后膜α1受体,能竞争性拮抗激动剂苯福林收缩血管升高血压的作用。能舒张静脉及小动脉,发挥中等偏强的降压作用。它与酚妥拉明不同,降压时并不加快心率,也少增加收缩力及血浆肾素活性,也能增加血中高密度脂蛋白(HDL)是浓度,减轻冠脉病变。
该药口服易吸收,2小时内血浓达峰值,生物利用度为60%,t1/2为2.5~4小时。但口服后降压作用可持续10小时,与血浆蛋白结合率达97%,在肝中广泛代谢,首关消除显著。
【临床应用及不良反应】适用于各型高血压,单用治疗轻、中度高血压,重度高血压合用β受体阻断药及利尿药可增强降压效。
不良反应有眩晕、疲乏、虚弱等,首次给药可致严重的体位性低血压,晕厥、心悸等,称“首剂现象”,在直立体位,饥饿、低盐时较易发生。将首次用减为0.5mg,并在临睡前服用,使可避免发生。
其他α1受体阻断药酮色林(凯他舍林ketanserin)兼有抗5-羟色胺S1受体的作用,也可有效地治疗高血压
α、β受体阻断药
拉贝洛尔
拉贝洛尔(labetalol)对α、β受体均有竞争性拮抗作用,其中,阻断β1、β2受体的作用程度相似,对α1受体作用较弱,对α2受体则无效,故负反馈调节仍然存在,用药后不引起心率加快作用。
本药降压作用温和,适用于治疗各型高血压,无严重不良反应,对梗塞早期,通过其降低心肌壁张力而产生有益的作用。静脉注射可治疗高血压危象。
六、作用于血管平滑肌的抗高血压药
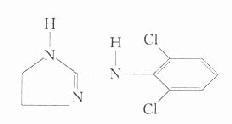
直接作用于血管平滑肌的抗高血压药肼屈嗪等,能直接松弛血管平滑肌,降低外周阻力,纠正血压上升所致的血流动力学异常。与其他类降压药不同的是,本类药物不抑制交感神经活性,不引起直立性低血压及阳萎等。久用后,其神经内分泌及植物神经的反射作用能抵消药物的降压作用(图26-3),从图可见最重要的反射变化是:①激活交感神经,致心输出量和心率增加而抵消降压作用,其增加心肌耗氧量的作用,对有严重冠状动脉功能不全或心脏储备力差者则易诱发心绞痛。②增强血浆肾素活性,肾素血症可增强交感活性导致循环中血管紧张素量增加而使血压上升,以上缺点必须合用利尿药及β受体阻断药加以纠正。
图26-3 久用扩血管药后其神经内分泌及植物神经的反射作用
直接作用于血管平滑肌的扩血管药可能作用于血管平滑肌细胞的兴奋――收缩偶联过程的不同部位,干予Ca2+的内流及Ca2+自胞内储库的释放,降低胞内游离Ca2+及其与平滑肌收缩蛋白的相互作用等。现知某些扩血管药可增加血管平滑肌的cGMP浓度,有的则通过开放钾通道使胞膜超极化而发挥作用。
肼屈嗪
肼屈嗪(hydralazine,肼苯哒嗪),为扩张小动脉的口服有效的降压药,对肾、冠状动脉及内脏血管的扩张作用大于对骨骼肌血管。适用于中度高血压,常与其他降压药合用。
口服吸收好,约65%~90%,给药1小时作用达峰值,维持约6小时。其不良反应有头痛、鼻充血、心悸、腹泻等。较严重时表现为心肌缺血和心衰。高剂量使用时可引起全身性红斑性狼疮样综合征,用量400mg/日,或更大,其发生率可达10%~20%,可见与剂量有关。将剂量降至200mg/日,上述反应少见。本经极少单用。
米诺地尔
米诺地尔(minoxidil,长压定),为作用强大的小动脉扩张药,口服吸收完全,能较持久地贮存于小动脉平滑肌中。其不良反应有水、钠潴留,心悸及多毛症,促进毛发生长可能与皮肤及毛发滤泡的血流增多有关。近证明本药激活了调节毛发杆蛋白的特殊基因而促进毛发杆的生长和成熟,故此药可作为治疗(男性)脱发药。
二氮嗪
二氮嗪(diazoxide,氯苯甲噻嗪),直接舒张血管平滑肌而降压,和米诺地尔一样,其降压机制部分是通过激活平滑肌细胞的IK(ATP)(ATP)所中介的钾通道,促进钾外流,使胞膜超极化,Ca2+通道失活,Ca2+内流减少。
临床上主要作静脉注射用,用于高血压危象及高血压脑病。不作长期用药。因此不良反应少见。如连用几天后,就应检测血糖水平。因本药可至高血糖症,此为药物激活了胰岛β细胞膜的IK(ATP),降低胰岛素释放所致。
钾通道开放剂吡那地尔(pinacidil),莱马卡林(lemakalim)为一类新型抗高血压药物,它们激活血管平滑肌细胞膜的IK(ATP)而发挥降压作用。
硝普钠
硝普钠(sodiumnitroprusside)又称亚硝基铁氰化钠(Na2Fe(CN)6NO・2H2O),属硝基扩张血管药,口服不吸收,需静脉滴注给药,起效快,约1分钟,停药5分钟内血压回升。其作用机制相似于硝酸酯类,能增加血管平滑肌细胞内cGMP水平而扩张血管。
用于高血压危象,特别对伴有急性心肌梗塞者或左室功能衰竭的严重高血压患者,治疗高血压危象一般按3μg/kg/分滴注,通过调整滴注速度来维持血压于所需水平。
由于该药能扩张动、静脉、降低前、后负荷而改善心功能,用于难治性心衰。
该药遇光易破坏,故滴注的药液应新鲜配制和避光。
不良反应有呕吐、出汗、头痛、心悸,均是过度降压所引起。本药毒性较你,在体内产生的CN-,在肝中被转化成SCN-,后者基本无毒,经肾排泄。但连用数日后,SCN-在体内蓄积,其浓度超过20mg/100ml时,可致中毒,因此宜监护SCN-的浓度。