�ڶ��ڡ���������ѧʵ�����
��������ѧ�������ﻯѧ�����Ϸ�չ�����ģ����о�����͵����ʽṹ�����ܵ��������ʵ�ѧ�ƣ��ں��ᡢ�����ʷ���ˮƽ�о���������ϡ����ƺ�Ԥ��Ļ��ơ����л��̣����������������飩��Ŀǰ��������ѧ�о��ȵ㣬��Щ�������Ը������������ͻ�����ʹ�����о�����ﵽ����ˮƽ�����о�������غͱ���ķ�����Ҳ�Ƿ���ˮƽ�о������������ơ�������Ϻͻ������Ƶķ�����ת����transformation����תȾ��ת����תλ������Ȼ�����������ڵķ�ʽ��Ҳ���˹��������鳣���õ��ֶΡ����������Ŀ��֮һ�ǻ����¡��gene clone���������¡������Ϊ��һ���ӻ���Ϊģ�������õ�����ģ����ӽṹ��ȫ��ͬ�Ļ���ʹ��Ҫ�����о����������ӵ�Ŀ�Ļ������ڴ������������������ڷ�����
������������ϸ��������״�ı�Ĺ��̽�ת����transformation�������ɾ������Դ������ϸ���Ĺ��̽�תȾ��transfection�����������壨�ɾ���������Ŵ����ʴ�һ������������һ�������Ĺ��̽�ת����transduction����һ����һ������һ��ת�Ƶ���������һ���Ĺ��̽�תλ��transposition������Щ�ζ��Ļ����תλ�ӡ�
һ�����̵ij��ù���
��һ������
���壨Vector���ǰ���ԴDNA��Ŀ�Ļ���������ϸ����ʹ֮����������������Ĺ��ߡ�������������plasmid�����ɾ��塢����˿״�ɾ����ճ��ĩ��������ճ�����������ȡ�������������Ҹ��ƣ��п�ѡ��ģ�����ɸѡ���������Ŵ���ǣ��й���ԴDNA�����λ�㣻�������С��������
���������ڶ���ϸ������Ⱦɫ�壨�ˣ�����Ķ����Ŵ����ӣ���˫����״DNA��ɣ�������ȫ��¶�������е����ʽ�ϡ��������Ͻ��ͺ��ɳ���֮�֡��Ͻ�����DNA���ø���ƣ�һ��ϸ���ɸ���1-5�����������ɳ�����DNA���ø���ƣ�һ��ϸ���ɸ���30-50���������������ù�ؿ���ֹ�����ʺϳɣ�ʹ������Ч����ԭ�ϣ����Ƹ����������������������Ʒ�ַ��࣬���õ���pBR322��pUCϵ�еȡ���Щ���������ж�����������縴������������ԭ��Ori�������ڸ����������������ر�ǣ������S��ù��Apr�����Ļ���Tcr�ȣ����ϣ���������Dz����ӣ�E.coli LacZ���ȣ����ڻ����������ɸѡ�������ӣ����Dz�����Lac��ɫ���������Trp�ȣ���ת¼��ֹ���У����ڲ������Դ����ת¼�������������ϻ�����������������ø���е㣬���������λ�㣬�ֽл�������λ�㣬�����¡λ�㡣
�����ɾ��������е����ɾ���M13ϵͳ��˫���ɾ���ϵͳ���ɾ���Ӧ����Ӧ������ϸ�����ʹ�á�������������ص㣬����ѡ�����Ӧ�á�
����������ø
����ø�ǻ������鼼������ȱ�ٵĹ��ߡ���Ҫ������������ø������ø������ۺ�ø����ת¼ø������ø�ȡ�
����������ø�Т��ͺ͢���������DNA����ø֮�֣��������ϸ�ʶ��������У�����ʶ�������ض��ĺ����ᴦ�п�DNA˫������ͨ����ָ���Ǣ���������DNA����ø��ʶ����ĺ�������������ᣬ��������ת�Գơ��пڷֿ��˺�ճ�ˣ�����3�䣭OH��5�䣭Pĩ�ˡ�����øƷ�ֶ࣬ʹ��ʱӦע���¶ȡ�����Һ������һ��1��g DNA/2-5��λø���ȷ�Ӧ������
| ø | ʶ������ | �п� | |
| Alu �� | ��AGCT�� | ��AG CT�� | ����� |
| ��TCGA�� | ��TC GA�� | ƽ���п� | |
| Eco R1 | ��GAATTC�� | ��G AATTC�� | �������� |
| ��CTTAAG�� | ��CTTAA G�� | ճ���п� |
����ø��T4�ɾ���DNA����ø��T4�ɾ���RNA����ø����ϣ��DNA����ø�ȡ�DNA����ø������ƽ�ˣ�Ҳ����ճ�ˡ���Ӧ����Mg2+��ATP���ڣ�pH7.5-7.6�������¶�37�棬30�����»��������½��������ǵ�������DNa ���ȶ��Ժ�ճ��ĩ�˵��˻��¶ȣ�һ��ƽ��������20-25�棬ճ��������12�����ҡ�
�ۺ�ø��DNA�ۺ�ø����DNAΪģ��ϳ�DNA��ϣ��DNA�ۺ�ø��ϣ��DNA�ۺ�ø���Ƭ�Σ�Klenow��Ƭ�Σ���T4��T7�ɾ���DNA�ۺ�ø�ȣ���RNA�ۺ�ø����DNAΪģ��ϳ�RNA��T7��T3�ɾ���RNA�ۺ�ø������ת¼ø����RNAΪģ��ϳ�DNA����RNA�����з����⣬���ִ�ϣ��DNA�ۺ�ø���Taq DNA�ۺ�ø������ת¼���ԣ���
��ϣ��DNA�ۺ�ø�����5���3��ۺ�ø���Ժ�5���3�䣬3���5������ø���ԡ�KlenowƬ����DNA�ۺ�ø�ݲݸ˾�ø���ò�����һ����Ƭ�Σ���5���3��ۺ�ø��5���3������ø���ԣ���3���5������ø���ԡ�������ȱ�ڷ��루Nick translation������Ǻ��ᣬҲ������DNA���вⶨ����DNA���ȡ�
����ø��DNase��RNase������øS1�ȣ���ˮ����Ӧ��DNA��RNA������øS1�ɽ��ⵥ��DNA��RNA����������Ҳ�ɽ���˫�����ᡣ����������ȥds-cDNA�ϳ��в����ķ��л���
ĩ��ת��ø��Mg2+�����£�ѡ��3�䣭OH�˵���DNAΪ����ӳɺ����ᣬ��Co2+�����£�ѡ��3�䣭OH��˫��DNAΪ����ӳɺ����ᣬ�γɶ�ۺ�����β�������ں���ĩ�˱�Ǻͺ������ӵĻ������β������������
��������øȥ��5�䣭P���ɷ�ֹ������DNAƬ��5���P���������ռ��ϰ���Ӱ��DNA����֮������ӣ�һ���ü�������ø��������DNA��ȥ5���P���ţ�������ø������Ŀ�Ļ����5���P��������3���OH���ӣ���ͨ����������һ������ʹ��������ȫ���ӡ��÷���������������Ч�ʣ�ͼ18-1����
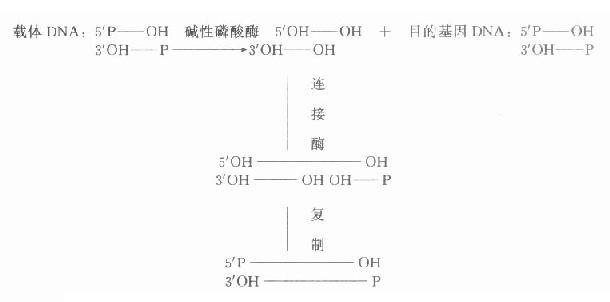
ͼ18-1������������ø����������DNA��Ŀ�Ļ���DNA������
����Ŀ�Ļ���
�������о��Ļ����ΪĿ�Ļ���������Ļ���ưл����ڻ����¡��������ʱ���߾���Ϊ���������ʱ���ߺ����������ԭ������Ŀ�Ļ���ɴ�ϸ������ֱ�ӷ���õ���������Ļ���ֲ���23��Ⱦɫ���ϣ����Ѵ�ֱ�ӷ��õ�����̵�Ŀ�Ļ�������˽�һ���ṹ��ͨ���˽������һ���ṹ���������ĺ��������л������˹��ϳɡ���������Ŀ�Ļ�����mRNA�ϳ�cDNA��complementaryDNA����ת¼DNA���õ���CDNAͨ�����ַ�ʽ���������ӣ���¡�ɵõ�ȫ��cDNA��Ƭ�Σ�����̽���Ʊ������з��������������о��������cDNAΪ�о����Ϸ�ӳ��mRNA��ת¼�����Ժ����Ӱ�����������ӳijһ����DNA�������ӵ��������ͼ18-2��
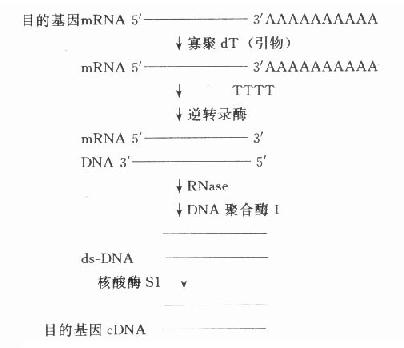
ͼ18-2��Ŀ�Ļ���cDNA�ĺϳ�
���������Ľ���
����������Ŀ����Ϊ�˱���Ŀ�Ļ���ı��桢�����ʹ���������������ù���ø��ͨ���������鼼����ת����ɸѡ��������Ҳ�ǻ����¡�Ĺ��̡�ʵ���������õ͵�������ϸ�����������ɾ���������죬��ֳ��ǿ���ص㣬����Ϊһ�ֺ�������ɸѡ�����壬������ȸߵ�����Ļ������Ƭ���ù���ø���룬���������У���ɸѡ����¡�õ�Ŀ�Ļ���������������������Ϊ���ڱ��棬ȡ�÷���Ļ���⡣����⽻�����о���֮�佻��Ŀ�Ļ���ij��÷�ʽ��
��һ����������
�������鷽���ܶ࣬�Ŀ���ͬһ������������ø�ֱ��������Ŀ�Ļ����г����Ӧ���пڣ��������ӡ�Ҳ����ĩ������ø�ϳ���������ͼ18-3�������꣬Okayama����Ϊʵ�����ձ���ã��÷����ɵõ�ȫ����cDNA��
������ת��
ת��Ŀ���ǰ��и�������������ϸ�������ƻ��Ե�Ŀ�Ļ���-����������װ��ϸ������ϣ������ʹĿ�Ļ�����������ϸ���ڸ��ơ�������ʵ�鲽��������£�
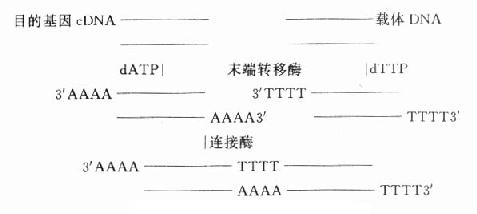
ͼ18-3���������鷽��֮һ
����ϣ���Ĵ���������ϸ��Ĥͨ�ԣ����ڻ������ϸ������ϣ����E.Coli cells��������Lb agar��������37������һ������ѡ3~5������䣬������50ml LB��������37�棬��ҡ����һ������A550�ⶨ��Ҫ��ϸ����ֳһ������һ��Ϊϸ�����������ڣ���Ұ���ͣ�rec����5x107cells/ml��Ϊ0.2-0.3��ȱ���ͣ�rec-��5x107cells/ml��Ϊ0.5-0.6��������ȥ����Һ����20ml��50mmol/L��CaCl2�������塣��ԡ20���ӣ��������ġ�������Һ����2.5ml��50mmol/l CaCl2������4��ɱ���48Сʱ������ת������competent cells��
���������羭�������齨�������������������ȣ���ת�� ��competent cells��������BRL��˾DH10B����Ϊ�����ñ�ˮԡ��ͬʱ��Falcon2059������ϵ���ȶ��������Թ��ñ�ˮԡ��ȡ100��l��Һ��DH10B����2059�Թ��У���10��ϡ�͵��ϻ��Ҵ�1��l����ԡ��ҡ2���ӣ�����10���ӡ�ȡ0.1-50ng���������Թܣ�����ҡ�ȣ���ԡ30���ӡ��˼䱸��42��ˮԡ������SOC��������42�汸�á����Թ�����42�棬��ҡ45���ӣ������ñ�ԡ2���ӡ�ʹcompetent cells����������������������塣��42��SOC������0.9ml���Թܣ�37������1Сʱ��1000rpm����10���ӣ�������Һ����200��lSOC�������������塣������LB-���S��ù�أ�100U/ml��������37������һ������ת���ľ�����γɾ��䣨���������п����S��ù�ػ������˽�Ŀ�Ļ���������Ļ����϶Ծ�����м���������LB����Һ�����������������Ľ�����ת����ɸѡ�������������Ĺ��̾��ǻ����¡�Ĺ��̡�
LB��������1L����10g�����ˣ�5g��ĸ�࣬10gNaCl����ѹ�����pH7.5��
SOB��������1L����20g�����ˣ�5g��ĸ�࣬0.5gNaCl����ѹ�����������2mol/Lþ��Һ��1mol/L�Ȼ�þ��1mol/L����þ�������ȣ����������������ǰ��1ml��100ml��������
SOC��������100ml��������ǰ����1ml���������2mol/L�����ǡ�
�ġ�����ķ����봿��
��������ڶ���ϸ�����粡����ϸ���������桢��ֲ��ϸ�������ֱ걾�У���ѪҺ����֯����Һ����Һ��������Դ�ı걾����˷��뷽�����Ӷ��������������DNA��RNA�ķ�Ȳ��죬���ַ��������Ժ���Ĵ���������Ҫ���в��죬�����ʵ��ǰӦ�Բ��õķ��������˽��ѡ���ܵ�˵������ķ����봿�������ܽ�ϸ���Ļ����ϣ����ñ��ӵ��л��ܼ����ᣨ�������ڻ���Һ����ˮ�ࣩ�����룬�������Ҵ�����ͪ���л��ܼ��������ջ��ܽ�ϸ���ķ�����걾��ͬ����ͬ������SDS��NaOH�����õ���ø���е��ó���������ȷ�����������ȡ��Ҫʹ�����ʱ��Գ������л��࣬�����ᱣ����ˮ�࣬�ﵽ��������Ŀ�ġ�ʵ��������걾�к������ľ��ǵ����ʡ�Ϊ�˳�ȥ��������в������л��ܼ������õķ����Ǽ����Ҵ����γ������ᣬͨ�����Ļ��պ��ᣬȻ����70��-80���Ҵ�ϴ�ӳ�������ȥ������Σ�����Ӱ������ܽ�����ƺ��������ø�ٷ�Ӧ��Ϊ�˵õ����ĺ�����õ���ø��ȥ���ף���RNAø��ȥRNA���õ�����DNA����DNAø��ȥDNA�����RNA��Ŀǰ������������Ʒ���ĺ�����������ɼ����ٵط���õ����Ⱥܸߵ�DNA��RNA�������ԭ���е����ú���ķ��������죬�е���������������ص����������Խ�ϴﵽ���롢���յ�Ŀ�ġ�
��һ�������ķ����봿��
��������E. Coli cells��LB����Һ250ml��������������50ml���Ĺܣ�4�棬3000rpm������15���ӡ���ȥ����Һ������ȥҺ����ǰӦ������������������Ⱦ����������lysis buffer(50mmol/L Glucose��10mmol/l EDTA��25mmol/l Tris-HCl��pH8.0)1-2mlʹ֮��������������5���ӣ���3.5-7ml������SDS/NaOH��Һ��1mol/l NaOH 8ml��20��SDs2ml������ˮ30ml������ҡ�ȣ��ñ�ˮԡ5���ӡ����û�����������DNA���Ӷ��ѣ�����2.5mol/L����ػ���Һ��pH4.8��2.6-5.2ml������ҡ�ȣ���ԡ5���ӡ�4�棬3000rpm������15���ӡ����������ʡ�ȡ����Һ������ˮ�Ҵ�12-24ml������Һ�Ķ�����������15���Ӻ�10000rpm���ģ�������Һ����ԼΪ�����������0.4��TE��10mmol/lTris-Hcl pH8.0��10mmol/l EDTA���ܽ������0.2����7.5mol/L����李����û��������ȣ��ñ�ԡ10���ӡ�4�棬10000rpm����5min��������Һ���ӳ�����0.4����10mmol/lTris-HCl pH7.5��10mmol/l EDTA����Һ��1/10�����5mg/ml RNase��37�棬30���ӱ��¡��ӵ�����phenol���ӣ�/CIAA(480ml�ȷ£�20ml���촼)�����ȡ�4�棬10000rpm������5���ӡ�ȡ�ϲ�Һ�ӷ�/CIAA�ظ�һ�Ρ�ȡ�ϲ�Һ��1/10���5mol/l NaCl��2����ˮ�Ҵ����D20���ҹ��D70��2Сʱ���ϡ�4�棬10000rpm������10���ӣ�������Һ����80���Ҵ���ҡ����ֱ��10000rpm���ģ�������Һ����ո����������Լ200��l��TE�ܽ⡣�ⶨ�������á�
����������������Ŀ�Ļ���ķ����봿��
��ȡ��������������������������������ø�г�Ŀ�Ļ�����Ӿ���룬ȷ�ϡ���200��l��2mg DNA������������Ϊ����ȡ10��l��100��g DNA����90��l TE����1��gDNA��2-5U����������ø��1/10�������Һ��1-2Сʱ���¡�����Һ�����ø��Ӧ�¶�������ø������졣1/10���3mol/l NaAc��2����ˮ�Ҵ�����70�棬2Сʱ����20���ҹ����10000rpm��10�������ģ�������Һ���Ҵ�������������TE�ܽ�������жϵ�DNA������Ŀ�Ļ����������Ӿ���루����Ӧ������DNA��ͬʱ��Ӿ������������¹۲��������DNA�������Ƚϣ�ȷ��Ŀ�Ļ��������ø���и�Ч����
����Ӿ������
| �����Ʊ��� | 1����֬������ | agarose(mg) | X50TAE(ml) | EB(�廯�Ҷ���l) |
| (agarose) | 1000 | 2.0 | 4.0 | |
| �������ml�� | ||||
| 100 |
| ��Ũ�ȣ������� | 0.3 | 0.6 | 0.7 | 0.9 | 1.2 | 1.5 | 2.0 |
| DNA���ȣ�kb���� | 60-5 | 20-1 | 10-0.8 | 7-0.5 | 6-0.4 | 4-0.2 | 3-0.1 |
| ��Ӿ����Һ�� | X50TAE(ml) | EB(��l) | �����(ml) | ||||
| 20 | 30 | 1000 |
X50TAE��Trismabase 54g������57.1ml��0.5m EDTApH8.0 20ml������ˮ��1000ml��
EB��10mg/mlethidium bromide��(EB���°��ԣ�����ӦС��)��
����Ӿȷ�Ϻ�ȡ����DNA������������ͬ�������п�����1.5�����۵㣨��65���۽⣩��֬����������Ӿ���롣��������£��г���Ŀ�Ļ������������������������Ĺ��У���ȡ������
���ӵ��۵�������ȡ������DNAƬ�� �������������ȵ�TE��10mmol/lTris-HCl pH8.0��0.1mmol/l EDTA������65��ˮԡ5���ӱ��£�ʹ������ȫ�ܽ⡣���������£��ӵ����ӣ�TE���ͣ�TE�����ϲ㣬ȡ�²�ӣ���������ȣ����û��ȣ���12000rpm��3�������ġ�����1-2�Ρ�ȡ�ϲ�Һ����0.1���3mol/L�����ƣ�pH5.2����2.5�������ˮ�Ҵ��������Ҵ���������������DNA������TE�ܽ⣬�ⶨ���������ã�������Ŀ�Ļ���ṹ������̽���Ʊ��ȣ��������۵��������շ��⣬����һ�����������������ݵ�Ӿ�����ĹܵͲ��Ӳ����ޣ��������ĵȷ�������DNAƬ�Ρ�
�������걾DNA�ķ����봿��
��5-10ml 1xNTE(NaCl 100mmol/L ,Tris-HCl10mmol/l pH7.4��EDTa 1mmol/L pH8.0)����ϸ��Ϊ2��107������֯Ӧ������Һ�����������ٽ��гɷ�ĩ���뻺��Һ������1/10-1/20�����V��10mg/ml����ø��Sigma ���ͣ���1/20v 10��SDS��37��2Сʱ���ӵ�����/3xNTE(3xNTE�Ϸ�)���ȣ�����7���ӣ���4�棬3000rpm��10���ӡ�ȡ����Һ����2mlTE(Tris-HCl 10mmol/L pH7.5��EDTa 1mmol/L pH8.0)����ֻ��ȡ��Ҵ�������2mlTE�ܽ��������1/30xNTE������øK(10mg/ml)���浰��ø�ظ�2-4���衣�ⶨ������
���ģ���ƷRNA�ķ��룬����
��106��ϸ��Һ�ӵ���������Һ[4mol/L�һ��������Σ�25mmol/L������pH7.0��0.5�������ᣨsarcosyl����0.1mol/l 2-�ϻ��Ҵ�]�������Ϊϸ������4-5�������ȡ���0.1���2ml/L�����ƣ�pH4.1����1����ӣ�����ˮ�Ϸ⣩��0.2���CIAA�����������һ��10���ӡ���ˮԡ15���ӡ�10000xg4�棬20���ӡ����ģ�����ɲ������С��ȡ������Һ���ӵ�����ͪ����2���Ҵ�������20�棬60�������ϡ�10000xg��4�棬20�������ġ�������Һ����1.5ml���Ĺ�Լ��0.3ml������Һ���ظ�3)���裬����10���ӣ�������Һ����75���Ҵ���10000xg��4�棬10�������ġ�������Һ����ո���15���ӣ����á�
�塢̽���Ʊ�
̽���Ʊ�����Ŀ�Ļ���ı�ǣ������Ƿ������õ���ȱ��ƽ�Ʒ�����������ĩ�˱�Ƿ��ȡ���������з�����Ԫ�أ���32P�ȣ��ͷǷ��������ʣ��������ء��ظ����ȣ���32P����õĺ�������ͬλ�أ�����ǵ�dNTP�����ʹ���������ţ����ڱ�ǣ��ص��DZȻ��Ըߣ��ɴ�9000Ci/mmol������Ħ����������ߣ��ɴ�1.70MeV��������ǵ�̽������Ӱʱ��̣������ȸߡ�32P�İ�����Ϊ14�죬Ӧ������ã�һ���Ǻ���һ����ʹ�ã�����������㣬����ʹ�ú�����ﴦ��������ѹ����ʹ��32P�����Ӧע�����������ʱӦ��1-1.5cm��ۼ���ϩ������л��������뱣��������ֱ�����䡣������Ա��ǰӦ������˼����������ڼ�⡣����ʵ���Ӧ��ר��̽�������Ǹ�-���ռ����������鹤�������֡��·��ȣ�������Ⱦ����������ͬλ�ر���ͬ��Ӧע������߷�����
�Ƿ����Ա����ø��ͻ�ѧ���Ƿ���ø�귽�������߲ⶨELISA�������ƣ�ֻ�DZ���ǵĺ�������˱���ǵĿ��壬��ʵ�ϱ���ǵĿ���Ҳ��Ϊ̽�룬�Ķ�����ʱӦ����ע�⡣����������Ʒ�������أ�biotin�����ظ�����ǵģ���������-dUTP��������-dATP���ظ���-dUTP�ȡ�Ѫ���أ�avidin�����������зdz��ߵ����ԣ���Ѫ���ر���Ϲ�������ø���������ø��Ѫ����-ø�������ӽ���Ӧ�����γɣ�̽��-������-Ѫ����-ø�����ABC������ø��������ɫ���۲�������һ���ø��Ӧ���ﲻͬ��ABC��������ɫ��Ϊ������Ա�۲�����ø��Ƿ����ӡ��ظ��Բ�ɱ��ߣ����������䱣�棬����������������൱����ѧ�����е�Ҳ��������Ӧø�꿹���γ����츴��������������൱���е�����Է��⡣��ѧ��Ƿ����ɱ��ͣ�����������Խϵ͡�����18-1��
��18-1��̽���Ǽ�����
| ����� | ��ⷽ�� | �ص� |
| 32P | �����ߣ�����Ӱ | ��������ߣ�������14�죬����ǿ��1.7MeV |
| 35S | �����ߣ�����Ӱ | �����ȸߣ��ֱ��ʺã�������87�죬0.17MeV |
| 3H | �����ߣ�����Ӱ | �����ȸߣ��ֱ��ʸߣ�������12�꣬0.01MeV |
| 125I | �����ߣ�����Ӱ | �����ȸߣ��ֱ��ʸߣ�������60�죬0.04MeV |
| ������ | ø��Ѫ���أ���ɫ | �����Ⱥã���������Դ�������ر걾 |
| �ظ��� | ø��ظ������壬��ɫ | �����Ⱥã��ֱ���һ�� |
| T-T������ | ø�꿹�����忹�壬��ɫ | �����Ⱥã����������γɶ��������� |
| BrdU | ø�꿹BrdU���壬��ɫ | �����Ⱥã�ϸ���ں��������Ʋ��� |
| ��-����֬�� | ӫ�� | ������һ�� |
ȱ��ƽ�Ʊ�Ƿ�����DNase ����DNA˫�����г�����пڣ�DNA�ۺ�ø����ȱ��ˮ��5��˺������3��������뱻��Ǻ�����ͬʱ���У��п�ƽ�����ƣ��ɾ�һ���DNA������ͼ18-4��5��
ȱ��ƽ�Ʒ����١���㡢�ɱ���Խϵ͡��Ȼ��Խϸߡ���Ǿ�һ����Ҫ������DNA�ۺ�ø����DNA�Ĺ��ܣ����ڴ����DNA��ǣ���1000bp��ã�������DNA��RNA�����ø÷���ǡ����������������ŵ㣬�ɴ���ȱ��ƽ�Ʒ��������С����˫DNA���ɱ�ǣ���Ǿ��ȣ�����ʸߣ������ܱ�ǻ�״DNA������ĩ��ת��ø�ɽ��С�β�ꡱ��β�������ڹѺ�����̽���ǣ����ڴ���Ӻ�������β�Ͷ̶���DZȻ��Խ��͡�β�겻����dTTP��ǣ���mRNA�Ķ�ۣ�A��β��Ӱ���ӽ������ԡ��Ѻ�����̽������ں��ᡰ�㡱ͻ���⣬��̽����ú���ϳ����˹��ϳɡ���¡̽��һ��ϳ���������
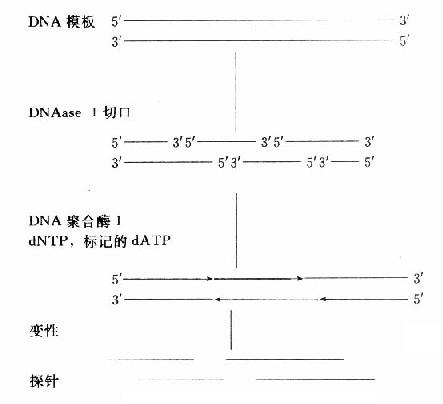
ͼ18-5��DNA��������Ƿ�
�ã���������ӽ�����ź�ǿ���ϳ�̽�볤�ȶ̣�һ����20-50������֮�䣬�����ϳɳɱ��ߣ����׳��־ۺ�ø�ϳɴ����ӽ�ʱ�䳤��̫�����������½����ϳ�̽����ƻ�Ӧע�������G��C��40��-60����һ�ּ�������ظ�������4����������������ӽ���������Ҫע��̽������������Ӧ��������������������С��ṹ��Ӱ���ӽ���һ���õ�̽��������ʵ���в��ܼ���ȷ�ϡ����⣬��������M13�ɾ�������ɸ��Ƶ���DNA���ص㣬���ƺϳ�DNA̽�룬����ת¼���壨DNA����ת¼�ϳ�RNA̽�롣�����GIBCoBRL��˾��Bethesda Research Laboratories��������ȱ��ƽ�Ʒ�����������Լ��У�bionick labeling system��Ϊ���������£�
| �Լ��� | 10x dNTP Mix.250��l | 10x Enzyme Mix 250��l |
| 0.2mmol/L each dCTP��dCTP��dTTP | 0.5 units/��l DNA Polymerase �� | |
| 0.1mmol/L dATP | 0.0075 units/��l DNase �� | |
| 0.1mmol/L biotin-14-dATP | 50mmol/L Tris-HCl(pH7.5) | |
| 500mmol/L Tris-HCl(pH7.8) | 5mmol/L magnesium acetate | |
| 50mmol/L MgCl2 | 1mmol/L ��-mercaptoethanol | |
| 100mmol/L ��-mercaptoethanol | 0.1mmol/L phenylmethyl- | |
| sulfonyl fluoride | ||
| 100��g/ml nuclease-free BSA | 50����v/v��glycerol | |
| 100��g/ml nuclease-free BSA | ||
| Control DNA��5��g pBR322 in TE | ||
| stop buffer 300 mM EDTA | ||
| autoclaved H2O |
�������裺
��5��l 10x dNTP Mix.������l DNA���൱1��g���ǵ�DNA����������l�������ˮ����45��l���ټ�5��l10x Enzyme Mix.���μ���1.5-2ml���Ĺܺ�15000xg����15���ӡ�16�汣��1Сʱ����5��l Stop Buffer��ֹ��Ӧ���Ҵ����������á�